Great expectations and generous reading of guidelines underestimate potential risk in oversight of COVID-19 pro-vaccine quality, safety testing, and manufacturing: 248 questions for FDA
David Wiseman, 1 L Maria Gutschi 2
1 Synechion, Inc., Dallas, TX 75252
ORCID: 0000-0002-8367-6158
2 Pharmacy Consultant, Ottawa, Ontario, Canada
ORCID: 0009-0008-0020-0826
Version: 091325
Funding: No funding was received for this study.
Conflict of interest: DW has acted as an expert witness or consultant in several legal cases related to parental authority regarding COVID-19 vaccines or their mandatory use.
Data availability: This is a review of the published medical and scientific literature, including regulatory documents released publicly of through FOIA requests.
ABSTRACT
Background
On December 6, 2023, Florida Surgeon General Ladapo wrote to the then FDA Commissioner and CDC Director Cohen of CDC, raising concerns about the COVID-19 pro-vaccines, particularly related to DNA contamination and the presence, in the Pfizer product, of an SV40 promotor-enhancer-ori sequence. These findings were confirmed by a number of labs, by regulators, or documents obtained under the Freedom of Information Act.
A response was furnished by FDA, which formed the basis for subsequent responses to questions regarding DNA contamination and other manufacturing and safety testing issues, including frameshift proteins.
Method
This document is written in the form of a point-by-point critique to this response, with several additional issues identified. Along with explanatory text, this document contains 248 questions to FDA.
Findings
The safety standard for an EUA requires that “the known and potential benefits of the product, […] outweigh [its] known and potential risks. “ Uncertainties, irregularities, or deviations from normal accepted practice in the manufacturing process represent “potential risks” FDA was required to consider. It was not necessary to prove that DNA contamination, the SV40 sequence, or frameshifted proteins did caused harm, the mere fact of these phenomena constituted an uninvestigated potential risk FDA was legally required to consider when granting the EUAs.
Given the rapid launch of the COVID-19 pro-vaccines, manufacturing and safety testing standards needed to be tightened, not loosened as described here, adding potential, if not actual risk.
Generous interpretation of various guidelines, and the word “expects” has been used by both sponsors and regulators to justify why a certain test should not be done or a safeguard implemented, without providing detailed justification supporting the assertion.
We detail a number of inconsistencies with statements made by regulators and data obtained from public records, in one case we found an instance of an undeclared (by regulators) genotoxicity study.
The developmental and regulatory failure that the findings related to residual DNA and frameshift proteins represent, are hardly evidence that these modRNA vaccines meet “ rigorous scientific standards,” and will no doubt erode further the trust in public health institutions.
Conclusion
We hope that our questions will allow us to understand the potential harms associated with the modRNA pro-vaccines in a way that begins to restore the public trust in the scientific process and health institutions, by engaging in a process of introspection and improvement of regulatory processes and decision-making.
These expedited and still experimental vaccines are the most complicated medical products ever deployed. Pfizer’s recently retired head of vaccine research, Dr. Kathrin Jansen, was quoted in Nature as saying ““We flew the aeroplane while we were still building it.” It is now time to ground the plane pending the answers to these, and no doubt, many more questions.
Keywords: Vaccine, COVID-19, Pfizer-BioNTech, Janssen, Moderna, vaccine safety, vaccine manufacturing, mRNA vaccine, DNA contamination, FDA, CDC, genotoxicity, mutagenicity
Table of Contents
2.1. Off-label claims related to serious outcomes . 5
2.2. Off-label claims related to pregnancy and lactation . 6
3. “Context of the manufacturing process” . 9
3.1. Manufacturing process changes – what was disclosed and what was promised? . 9
3.2. Was the descriptive analysis performed? . 12
3.3. EMA concerns regarding the Pfizer process and the process change . 13
3.4. EMA concerns regarding residual DNA . 14
3.6. Formulation and process changes after nonclinical studies . 17
4. Why are the possible harms from residual DNA? . 18
4.1. Prior downgrading of integration risk for residual DNA . 18
4.2. Risk of chromosomal integration: nuclear access of DNA .. 19
4.3. Additive risks of integration by residual DNA and by DNA reverse transcribed from modRNA . 22
4.3.1. “the mRNA is destroyed” . 22
4.3.2. “the RNA can't get into the nucleus, the nucleus has a one-way valve” . 22
4.3.3. “our cells do not have the enzyme necessary to get RNA back into DNA” . 23
4.4. The need for integration studies . 24
4.5. Risk of episomal / extrachromosomal expression . 26
4.6. Non-integrating mechanisms of DNA toxicology . 26
5. Experimental findings regarding residual DNA . 27
6. What are the guidelines concerning residual DNA? . 28
6.1. Guidelines on amount of residual DNA . 28
6.2 Guidelines on additional considerations for amount of residual DNA . 29
6.3. Guidelines on measurement of residual DNA . 30
6.4. Guidelines on fragment size of residual DNA . 31
6.4.1. What do the guidelines say about fragment size of residual DNA? . 31
6.4.2. Is fragment size of residual DNA being measured? . 32
6.4.3. Fragments of residual DNA exceeding the 200 bp guideline have been reported . 33
6.4.4. Concerns regarding residual DNA fragments smaller than the 200 bp guideline . 34
7. Concerns regarding intact plasmid sequence elements . 35
7.1. Antibiotic resistance gene . 35
7.2. SV40 promoter-enhancer and other sequences . 36
7.2.1. Concerns about specific SV40 sequences and SV40 proteins must not be conflated . 36
7.2.2. Possible consequences of the SV40 enhancer-promotor-ori sequences . 36
7.2.3. Was the SV40 and other sequence and other sequences identified to FDA? . 36
7.3. Unintended sequences in DNA plasmid vector 39
8. RNA or DNA Lipid Adducts, “Process 3” . 40
8.1. Formation of RNA or DNA Lipid Adducts . 40
8.2. Process 3: Pfizer buffer change . 42
9. Novel heterotrimers in bivalent pro-vaccines: Process 2 for Moderna, Process 4 for Pfizer 43
10. Safety studies with modRNA “and lipid nanoparticle together that constitute the vaccine” ? . 44
10.1. Which formulations were used in Moderna’s nonclinical safety studies? . 44
10.1.1. Was the modRNA that “constitutes the vaccine” used in genotoxicity studies? . 48
10.1.3. Was LNP size comparable in Moderna’s safety studies? . 49
10.1.4. Other differences in manufacturing and formulation of mRNA 1647 used in the PK study . 50
10.1.5. Inadequate preclinical use of animals with spike-ACE2R binding relevant characteristics . 51
11. Inadequate studies failed to predict modRNA and spike protein PK and expression kinetics . 52
11.1. Discrepancies between messaging and data on modRNA, spike persistence and distribution . 52
11.2.1. Unsupported expectations about modRNA and protein metabolism .. 53
11.2.2. Unsupported expectations about local distribution and action . 54
11.2.3. Unsupported expectation about LNP and mRNA kinetics . 54
11.3. Failure to follow guideline provision for case-by-case consideration . 55
11.4. Superficial analysis of limited biodistribution studies, lacking follow-up: Moderna . 56
11.4.1. Quality and methodology issues . 57
11.4.2. Evidence for mRNA dependent distribution . 57
11.5. Superficial analysis of limited biodistribution studies, lacking follow-up: Pfizer 60
11.6. Biodistribution studies: summary . 61
12. Safety studies: preclinical genotoxicity . 61
12.2. What genotoxicity tests were performed on COMIRNATY or SPIKEVAX? . 63
12.3. What genotoxicity tests were performed on ingredients related modRNA products? . 64
12.3.1. Relevance and reliance on supportive studies involving related modRNA products . 64
12.3.3. Missing genotoxicity studies of SPIKEVAX ingredients or related products? . 65
12.3.4. Questions regarding nonclinical studies other than genotoxicity . 68
14. modRNA vaccines elicit cryptic uncharacterized frameshift proteins of unknown toxicity . 72
15. COVID-19 pro-vaccines meet FDA definition of gene therapy . 76
17. Topics for further discussion . 79
1. Introduction
On December 6, 2023, Florida Surgeon General Ladapo wrote (1) to the then FDA Commissioner Califf and CDC Director Cohen, raising concerns about the COVID-19 pro-vaccines, particularly related to DNA contamination and the presence, in the Pfizer product, of an SV40 promotor-enhancer-ori sequence, reported originally by McKernan et al. (2) and expanded upon in a then recent preprint. (3)
A response was furnished on December 14, 2023, (4) by then CBER Director Marks. Rather than allaying his concerns, Dr. Ladapo believed that this response served only to heighten them, prompting Dr, Ladapo to call for a halt in the use of COVID-19 mRNA Vaccines. (5)
Dr. Marks’ response has formed the basis for subsequent responses to questions regarding DNA contamination since then, despite the confirmation of findings of DNA contamination that exceeded guidelines, and the presence of the SV40 sequences in the Pfizer product. This confirmation has been furnished by laboratories around the world (6-10) and, notably, by high school intern students working under FDA supervision. (11) Further, in numerous FOIA disclosures around the world, regulators have confirmed the presence of the SV40 sequence (including the Marks response (4) ) and the fact that Pfizer had chosen not to specifically disclose this regulatory sequence. Our own unpublished work has confirmed this with the Pfizer 2024-2025 JN.1 formula.
Since the Marks response has formed the basis for FDA’s position on this topic, this document is written in the form of a point-by-point challenge, along with additional points related to safety testing or manufacturing of these products.
Comments are provided on a consolidated point-by-point basis, raising questions in text and provided in list format the end of this document. While FDA is not bound by the actions of regulatory agencies in other countries, by FDA’s citing WHO guidelines as well as “internationally agreed upon recommendations” (4) we are justified in also citing the deliberations of other regulators to inform our expectations of FDA’s actions. Further, in many instances, FDA staff have played a major role in the drafting of various WHO guidelines.
The term pro-vaccine is used here. Unlike conventional vaccines, the modRNA products do not contain target antigens. Rather they contain the genetic instructions that are read by a patient’s body to produce the target spike protein antigen. This is somewhat analogous to the activation of pro-drugs, molecules that lack the desired pharmacologic action, but that are converted by the body to an active form. (12) We employ the term “pro-vaccine” (13, 14) to signify this important distinction, which again has regulatory consequences.
2. Misinformation
2.1. Off-label claims related to serious outcomes
Dr. Marks states: “Given the dramatic reduction in the risk of death, hospitalization and serious illness afforded by the vaccines, lower vaccine uptake is contributing to the continued death and serious illness toll of COVID-19.”
The indications stated at the time of Dr. Marks’ response in the package inserts for COMIRNATY and SPIKEVAX read:
“COMIRNATY is a vaccine indicated for active immunization to prevent coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in individuals 12 years of age and older.” (15) (and more recently (16) )
“SPIKEVAX is a vaccine indicated for active immunization to prevent coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in individuals 12 years of age and older.” (17) (and more recently (18) )
Question 1: Please confirm that neither label includes the indication that the vaccine reduces“the risk of death, hospitalization and serious illness” of COVID-19 disease. Q1
Question 2: Has FDA reviewed data that meet the standard of “substantial evidence” “consisting of adequate and well-controlled investigations” and support the claim that either vaccine reduces “the risk of death, hospitalization and serious illness” of COVID-19 disease. Please provide. Q2
Question 3: If FDA considers the data it has reviewed as supporting the claim that either vaccine reduces “the risk of death, hospitalization and serious illness” of COVID-19 disease and meeting the standard of “substantial evidence” “consisting of adequate and well-controlled investigations,” please state whether they do so according to the evidentiary standards set forth in FDA’s 1998 guidance[1](19) or or according to later 2019 (20) and 2023 (21) documents that expand the scope of the types of data that can be used to support certain claims or expanded indications. Q3
Question 4: If FDA has reviewed data that do not meet the standard of “substantial evidence” “consisting of adequate and well-controlled investigations” but purports to support this claim, please provide the data and state the evidentiary standard such data meet, if at all. Q4
Question 5: If FDA has reviewed data that do not meet the standard of “substantial evidence” “consisting of adequate and well-controlled investigations” please describe the deficiencies in the data that preclude the inclusion of language asserting that the COVID-19 vaccines reduce “the risk of death, hospitalization and serious illness” of COVID-19 disease Q5
Question 6: Please advise whether, in the absence of data meeting the “substantial evidence” standard as well as authorization by FDA of a labeling change to include a claim that the vaccine reduces “the risk of death, hospitalization and serious illness” of COVID-19 disease, manufacturers making such a claim would be in violation of statues and regulations regarding off-label promotion? How is your answer influenced by FDA’s draft guidance on Scientific Information on Unapproved Uses (SIUU)? (22) Q6
Question 7: What is the regulatory status of Dr. Marks’ (4)statement regarding a“dramatic reduction in the risk of death, hospitalization and serious illness afforded by the vaccines”? Coming from Dr. Marks, a senior FDA official, does this represent an amendment to the approved labeling, a regulatory guidance, an enforcement policy, SIUU, or personal medical opinion? Q7
FDA’s draft guidance on SIUU (22) recommends that in order, inter alia , to facilitate a firm’s obligations to ensure that FDA-required labeling accurately reflects “what is known about the safety profile of the drug,” “to ensure that the FDA-required labeling is not false or misleading,” and to dispel the notion of a “ firm’s intent that the medical product be used for an unapproved use,” (p6/29) sponsors providing SIUU should include, inter alia , “A statement that the unapproved use(s) of the medical product has not been approved by FDA and that the safety and effectiveness of the medical product for the unapproved use(s) has not been established.” (p16/29)
If these sorts of safeguards on the dissemination of SIUU apply to sponsors, who by virtue of their commercial interests, would be suspected by the medical and lay community of what FDA calls “persuasive marketing techniques” (p18/29 in (22) ), then at least similar safeguards and the principles they embody should exist in the case of government health agencies about whom there would be no suspicion.
Question 8: Although this draft guidance applies to sponsors, given Dr. Marks’ statement concerning a “dramatic reduction in the risk of death, hospitalization and serious illness afforded by the vaccines,” and the absence of the corresponding claim in the package insert, is it FDA’s intent that the modRNA COVID-19 vaccines be used to reduce these serious outcomes? Q8
Question 9: Per Question 8, if it is FDA’s intent that the COVID-19 vaccines be used to reduce serious outcomes, would the issuance of such a statement on this use by FDA without the qualifying language concerning this unapproved use, misleadingly imply that this use had been approved by FDA? Q9
Question 10: Did the provision of EUA product to patients who were counseled that these products reduce the risk of death or serious outcomes, violate the provider agreement, which requires that the provider must confine representations to those consistent with the contents of the Fact Sheet - eg (23, 24) ? Q10
2.2. Off-label claims related to pregnancy and lactation
The past and current package inserts for COMIRNATY and SPIKEVAX state:
· “Available data on COMIRNATY administered to pregnant women are insufficient to inform vaccine-associated risks in pregnancy.” (15, 17)
· “It is not known whether COMIRNATY is excreted in human milk. Data are not available to assess the effects of COMIRNATY on the breastfed infant or on milk production/excretion.” (15)
· “Available data on SPIKEVAX administered to pregnant women are insufficient to inform vaccine-associated risks in pregnancy.” (17)
· “It is not known whether SPIKEVAX is excreted in human milk. Data are not available to assess the effects of SPIKEVAX on the breastfed infant or on milk production/excretion.” (17)
Similar language appears in the original versions released in 2021 (COMIRNATY) and 2022 (SPKEVAX).
Question 11: Please confirm that the above excerpts do appear in the respective package inserts. Q11
The CDC web site (25) stated:
· “Everyone ages 6 months and older is recommended to get the updated COVID-19 vaccine. This includes people who are pregnant, breastfeeding, trying to get pregnant now, or those who might become pregnant in the future.”
· “ Evidence shows that: COVID-19 vaccination during pregnancy is safe and effective.”
· “CDC recommendations align with those from professional medical organizations serving people who are pregnant, including the: American College of Obstetricians and Gynecologists , Society for Maternal Fetal Medicine , and American Society for Reproductive Medicine ”
Question 12: Has FDA reviewed data that meet the standard of “substantial evidence” “consisting of adequate and well-controlled investigations” and support the claim that either vaccine is safe and effective for use in pregnancy or lactation. Please provide. Q12
Question 13: If FDA considers the data it has reviewed as supporting the claim that either modRNA vaccine is safe and effective for use in pregnancy and meeting the standard of “substantial evidence” “consisting of adequate and well-controlled investigations,” please state whether they do so according to the evidentiary standards set forth in FDA’s 1998 guidance (19) or according to later 2019 (20) and 2023 (21) documents that expand the scope of the types of data that can be used to support certain claims or expanded indications. Q13
Question 14: If FDA has reviewed data that do not meet the standard of “substantial evidence” “consisting of adequate and well-controlled investigations” but purports to support this claim, please provide the data and state the evidentiary standard such data meet, if at all. Q14
Question 15: If FDA has reviewed data that do not meet the standard of “substantial evidence” “consisting of adequate and well-controlled investigations” please describe the deficiencies in the data that preclude the removal of tempering labeling language and/or the inclusion of language asserting that the COVID-19 vaccines are safe and effective in pregnancy and lactation. Q15
Question 16: Please advise whether, in the absence of data meeting the “substantial evidence” standard as well as authorization by FDA of a labeling change to include a claim that the vaccine is safe and effective in pregnancy, manufacturers making such a claim would be in violation of statues and regulations regarding off-label promotion? Q16
Question 17: Please confirm that CDC’s recommendation for use of the COVID-19 vaccines in pregnancy and lactation, along with CDC’s representation that “Evidenceshows that: COVID-19 vaccination during pregnancy is safe and effective,” is misleadingly inconsistent with the wording in the COMIRNATY and SPIKEVAX package inserts concerning insufficient data to inform vaccine-associated risks in pregnancy,whether the vaccines are excreted in breast milk, and the lack of data on the effects of the vaccines on the breastfed infant or on milk production/excretion. Q17
Language could not be identified on this CDC web page that refers to the language in the COMIRNATY and SPIKEVAX labelling concerning what is known about the risks of these products in pregnancy and lactation.
Question 18: Please confirm that the absence of prominent language detailing the risks of these products in pregnancy and lactation as described in FDA approved labeling, from CDC’s related recommendations, exacerbates the misleading nature of these recommendations. Q18
Question 19: Please provide the contents and URLs of all current FDA and CDC web pages that discuss the use of these products in pregnancy and lactation. Please detail what steps will be taken to ensure that prominent language will be placed, if currently absent, detailing the risks of these products in pregnancy and lactation as described in FDA approved labeling. Q19
A FDA web page “Facts about COVID-19” (26) appears to endorse CDC’s recommendation as seen in this screen shot. (from 2024)

This page linked to the CDC page cited above (25) as well as another CDC page (27) which described the safety and effectiveness of the COVID-19 vaccines not as “safe and effective” as above but only in terms of a “growing body of evidence … that the benefits of vaccination outweigh any potential risks of COVID-19 vaccination during pregnancy:”
“Staying up to date with COVID-19 vaccinations is recommended for people who are pregnant , trying to get pregnant now, or who might become pregnant in the future, and people who are breastfeeding. A growing body of evidence on the safety and effectiveness of COVID-19 vaccination indicates that the benefits of vaccination outweigh any potential risks of COVID-19 vaccination during pregnancy.” (original links preserved)
The current page states: (28)
“Studies including hundreds of thousands of people around the world show that COVID-19 vaccination before and during pregnancy is safe, effective, and beneficial to both the pregnant woman and the baby. The benefits of receiving a COVID-19 vaccine outweigh any potential risks of vaccination during pregnancy.
FDA’s page also linked to a 47 second video on FDA’s YouTube channel. [2] The video (screenshots below) is dated May 19 2022.
|
|
|
|
|
|
|
|





Figure 1 : Screenshots from May 19 2022 episode of “Just a minute with Dr. Peter Marks.”
YouTube’s “show transcript” feature reads: “ Pregnant or breastfeeding women can certainly receive a COVID-19 vaccine and should discuss the potential benefits and risks of vaccination with their healthcare provider. There is currently no evidence that any vaccines, including COVID-19 vaccines, cause fertility problems in either women or men. If you are pregnant or breastfeeding, or might become pregnant in the future, the CDC recommends getting vaccinated. ”
Question 20: Please confirm that FDA’s endorsement of CDC’s recommendation for use of the COVID-19 vaccines in pregnancy and lactation, along with CDC’s related representations described above is misleadingly inconsistent with the wording in the COMIRNATY and SPIKEVAX package inserts concerning insufficient data to inform vaccine-associated risks in pregnancy,whether the vaccines are excreted in breast milk, and the lack of data on the effects of the vaccines on the breastfed infant or on milk production/excretion. Q20
There is no language in this video or in the description provided on the YouTube site to alert the viewer to the language cited above appearing in the package inserts for the Pfizer and Moderna products.
To an unprofessional eye, FDA’s video appears well-produced with clear dialog, engaging graphics and an upbeat soundtrack. It is one of a series of over 40 short videos addressing mainly COVID-19 related topics and titled “Just a minute with Dr. Peter Marks.” The series, as its title indicates, develops a celebrity-like status for its featured interlocutor, a senior FDA official, as a means to add authority and credibility to the message, in this case FDA’s endorsement of a CDC recommendation concerning the use of COVID-19 vaccines in pregnancy and lactation.
FDA’s recent draft guidance on SIUU (p18/29 in (22) ) cited in preface to Question 8 frowns upon the use by sponsors to disseminate SIUU using “persuasive marketing techniques” that could include “the use of celebrity endorsements.” The effective creation and deployment of the Director of CBER as a celebrity persona to endorse CDC’s recommendation for a use characterized by the label as having insufficient data, heightens the concerns expressed in my preface to Question 8 that government agencies have a higher duty to conform to the principles embodied in FDA’s draft guidance on the dissemination of SIUU.
Question 21: Please confirm that the absence of prominent language detailing the risks of these products in pregnancy and lactation as described in FDA approved labeling, from FDA’s written and video endorsement of CDC’s related recommendations, exacerbates the misleading nature of both FDA’s endorsement and CDC’s recommendations. Q21
Question 22: Did the provision of EUA product to patients who were counseled that these products were safe and effective for use in pregnancy and lactation violate the provider agreement, which requires that the provider must confine representations to those consistent with the contents of the Fact Sheet - eg (23, 24) ? Q22
3. “Context of the manufacturing process”
Dr. Marks stated: “ Perpetuating references to this information about residual DNA without placing it within the context of the manufacturing process is misleading.”
We suggest that the statement be revised to read:
“ Perpetuating references to this information about residual DNA without placing it within the context of the change in manufacturing process is misleading.”
Here follows a number of questions about the change in the manufacturing process.
3.1. Manufacturing process changes – what was disclosed and what was promised?
Pfizer [3] published (29) the initial results from its pivotal trial of its COVID-19 vaccine in the NEJM on December 10, 2020, the same day the VRBPAC was convened to discuss its authorization. The protocol for the study was provided as an online supplement to the publication. The protocol Amendment 7, in the version dated October 6, 2020 (p131/376) “Added an additional exploratory objective to describe safety and immunogenicity in participants 16 to 55 years of age vaccinated with study intervention produced by manufacturing ‘Process 1’ or ‘Process 2.’
The protocol elaborates (p174/376): “The initial BNT162b2 was manufactured using “Process 1”, however “Process 2” was developed to support increased scale of manufacture. In the study, each lot of “Process 2” manufactured BNT162b2 will be administered to approximately 250 participants 16 to 55 years of age. The safety and immunogenicity of prophylactic BNT162b2 in individuals 16 to 55 years of age vaccinated with “Process 1” and each lot of “Process 2” study intervention will be described. A random sample of 250 participants from those vaccinated with study intervention by manufacturing “Process 1” will be selected for this descriptive analysis.”
It is now public knowledge that Pfizer’s Process 1 used a DNA template generated by a PCR method, with likely minimal plasmid involvement. Switching to a plasmid – E. coli -based Process 2 similar to the one used by Moderna introduced the possibility of a very different profile of potential impurities and contaminants. These included the starting burden of residual DNA as a process related impurity, the types of DNA (e.g. plasmid sequence elements such as promoters, enhancers, and antibiotic resistance genes), and DNA and endotoxin contaminants from the E. coli . Certainly EMA in their February 19 2021 assessment of the Pfizer product noted (p16/140) that this change “may result in a different impurity profile in the active substance” (30) The “comparability between clinical and commercial material” (p31/140) , along with other issues were the subject of Major Objections (MOs) that would have precluded authorization.
These sorts of process changes introduced a new risk profile to the drug product, that in non-pandemic circumstances would have triggered at least a risk assessment, and possibly an appropriately sized clinical comparability study. FDA’s own guidance entitled “Q5E Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process” (31) instructively provides some general principles by which comparability should be determined (emphasis added):
“A determination of comparability can be based on a combination of analytical testing, biological assays, and, in some cases, nonclinical and clinical data. If a manufacturer can provide assurance of comparability through analytical studies alone, nonclinical or clinical studies with the postchange product are not warranted. However, where the relationship between specific quality attributes and safety and efficacy has not been established , and differences between quality attributes of the pre- and postchange product are observed, it might be appropriate to include a combination of quality, nonclinical, and/or clinical studies in the comparability exercise.”
The emboldened clause certainly applies to the modRNA vaccines. Given the necessity for clinical data, to estimate the size of such a study, FDA’s June 2020 guidance “Development and Licensure of Vaccines to Prevent COVID-19” (32) is instructive: “The pre-licensure safety database for preventive vaccines for infectious diseases typically consists of at least 3,000 study participants vaccinated with the dosing regimen intended for licensure.”
This is a far cry from an analysis that is merely “ exploratory ,” “descriptive” and involves only 250 subjects per arm. Perhaps the discrepancy reflects FDA’s switch from the BLA approach described in the June 2020 guidance (32) to an EUA approach in the October guidance. (33) This switch lowered the evidentiary bar from the BLA standard of “established as safe and effective” based on “substantial evidence” “consisting of adequate and well-controlled investigations.” to the EUA standard of “believes may be effective” based on a “totality of evidence” not requiring clinical studies at all. [4]
Further, an inadequately characterized manufacturing change may have fallen foul of cGMP. This may have been circumvented since. in a public health emergency, the Secretary of the Department of Health and Human Services may “ authorize, with respect to an eligible product, deviations from current good manufacturing practice requirements.” [5] (34) This seems also to have been the approach of EMA who issued a time-limited exemption regarding certain GMP issues that were the subject of Major Objections. This exemption, along with data submitted by Pfizer and the imposition of time-bound “Specific Obligations” (binding conditions to the authorization) and “Recommendations” (not binding but important considerations for future development) allowed EMA to lift the Major Objections and issue its Conditional Market Authorization “ In view of the declared Public Health Emergency of International Concern.” (p32/140 in (30) )
If the Secretary of HHS did issue such a waiver covering matters relating to this sort of process change, VRBPAC members do not appear to have been fully informed either at the meeting of December 10, or the meeting of October 22, 2020 which introduced the EUA framework. FDA’s Dr. Jerry Weir described some of the differences between the cGMP requirements for full (BLA) licensure and the “expectations” under EUA. With the main differences relating to stability and lot release testing, Dr. Weir explained that FDA expected complete details and validation of the manufacturing process, (35) further stating that “ The CMC expectations are very similar for EUA use or licensure.” (p201/408 of transcript (36) ).
Given these representations, the fact of this critical manufacturing change as well as the details of proposed post-authorization requirements or undertakings would be significant components of the “totality of scientific evidence available” VRBPAC was asked to consider when answering FDA’s voting question on December 10, 2020 as to whether or not “the benefits of the Pfizer-BioNTech COVID-19 Vaccine outweigh its risks for use in individuals 16 years of age and older?”

Figure 2 : Voting question for December 10, 2020 VRBPAC meeting [6]
The absence of this information compromises VRBPAC’s ability to assess the benefits and risks associated with the manufacturing change, impugning the integrity and validity of their vote.
Guetzkow and Levi documented Pfizer’s manufacturing changes, relative to the clinical trial and public distribution batches in a Rapid Response published in the British Medical Journal. (37) They described differences in the numbers of adverse events reported to VAERS with various batches and highlighted the “ the importance of publicly disclosing the analysis comparing reactogenicity and safety of process 1 and 2 batches as specified in the trial protocol.”
Further aspects of possible GMP deviations in the use of a “GMP-like” standards ae discussed in section 3.5 . The following questions arise:
Question 23: Did the Secretary of HHS authorize deviations from cGMP regarding the COVID-19 vaccines? Was this a general waiver for all COVID-19 vaccines or for specific vaccines and/or specific issues? Did such a waiver cover any cGMP issues stemming from the Process 1 to Process 2 change for Pfizer’s product?Please provide a copy of all relevant cGMP waivers. Q23
Question 24: When did FDA first learn that Pfizer would be changing from Process 1 to Process 2? Q24
Question 25: After learning about Pfizer’s process change, did FDA consider this change to constitute, absent a comparability analysis, grounds for a non-approvable status or the issuance of something akin to the EMA Major Objection? Q25
Question 26: To what extent did the challenges related to Pfizer’s process change contribute to FDA’s change in regulatory approach from a BLA pathway described in the June 2020 guidance (32) to an EUA pathway describedthe October 2020 guidance (33)? Q26
Question 27: Within the BLA framework described in the June 2020 guidance (32) what comparability requirements did FDA place or would have placed on Pfizer related to the proposed change in manufacturing process? How would these requirements differ under an EUA framework? Q27
Question 28: What combination of provisions, akin to those adopted by EMA, such as GMP waivers, additional pre-EUA data provided by Pfizer, post-EUA obligations and commitments, did FDA make in order to obviate any delays in authorization or approval caused by the process change? Q28
Question 29: Absent these provisions, by how long would the issuance of Pfizer’s EUA have been delayed? Q29
Question 30: Relating to this process change, did FDA request a risk assessment from Prizer? Was one provided and when? Did FDA conduct its own risk assessment? Was any risk assessment addressing this issue, if one exists, disclosed to VRBPAC or publicly. If not please provide. Q30
Question 31: Please confirm that there is no reference to the Process 1 to Process 2 manufacturing change in the meeting materials for the VRBPAC meeting of December 10 2020. Was VRBPAC fully informed of the fact and details of the manufacturing change, including protocol Amendment 7, and if so when and in what form? Q31
Question 32: What was the regulatory basisfor authorizing a process change based on a descriptive comparability analysis involving 250 subjects per arm? Does this analysis meet the BLA “substantial evidence” or merely the EUA “totality of evidence standard”? If the answer is the latter, how is this lowered standard consistent with FDA’s representation in its October 6 2020 (33) guidance and to VRBPAC on October 22, 2020 (38) that it would still require data “from at least one well-designed Phase 3 clinical trial that demonstrates the vaccine’s safety and efficacy in a clear and compelling manner”? Q32
Question 33: Regarding the process change, was VRBPAC fully informed and educated about the lowering of the “substantial evidence” or “clear and compelling” standards to a “totality of evidence” standard? When? In what form? Q33
Question 34: Was VRBPAC fully informed and educated about the existence and details of any cGMP waivers? When and in what form? Other than the publication of Pfizer’s study and protocol in the NEJM[7] on the same day as the VRBPAC meeting, did VRBPAC members receive these materials prior to the December 10 2020 meeting? Q34
3.2. Was the descriptive analysis performed?
Information about the Process 1 to Process 2 change has been more forthcoming from the European Medicines Agency (EMA) than FDA. In its Assessment Report dated February 19 2021, (30) EMA (p34/140) notes the need to demonstrate the biological, chemical and physical comparability of Drug Substance (DS) and Drug Product (DP) produced by the two processes.
“The commercial batches are produced using a different process (Process 2), and the comparability of these processes relies on demonstration of comparable biological, chemical and physical characteristics of the active substance and finished product.”
In relation to this and other issues, EMA (p137/140) determined that “Given the emergency situation, it is considered that the identified uncertainties can be addressed post-authorisation in the context of a conditional MA [marketing authorization],” expecting the descriptive comparability analysis of clinical data in February 2021 ( p69/140).
A heavily redacted comparability report involving bench-top analytical and limited in vitro testing dated March 2021 was released by the Australian Therapeutic Goods Administration (TGA) in June 2022. (39) However the fate of the clinical descriptive study was not known publicly until the UK Medicines and Healthcare products Regulatory Agency’s (MHRA) provided a response to FOI request 23/510. (40)
MHRA explained the need for the descriptive clinical study a little differently from EMA. (30)
“Typically, such changes can be supported by analytical data; however, due to the nascent regulatory landscape for COVID-19 vaccines, in October 2020 an exploratory objective was added in the C4591001 study to describe safety and immunogenicity of vaccines produced by manufacturing “Process 1” or “Process 2” […] ”
Shockingly, MHRA revealed that “this process comparison was not conducted as part of the formal documentation within the protocol amendment.” The study objective that had been added in Amendment 7 in October 2020, had, according to MHRA, been removed in amendment 20 in September 2022 “due to the extensive usage of vaccines manufactured via “Process 2.” “
This episode raises several questions:
Question 35: How many different lots of Process 2 Drug Product (DP) were deployed in Pfizer’s pivotal trial C4591001? How many subjects received Process 2 DP (by lot number)? Q35
Question 36: Please confirm the information provided by MHRA in their FOI response 23/510 that the first subject to receive Process 2 DPdid so on October 18 2020. Please provide the date when the last subject to receive Process 2 DP did so. Q36
Question 37: Please confirm the information provided in EMA’s report (30) that the descriptive clinical comparability analysis was expected in February 2021. If this was not the case, what was the timeline for submission to FDA of Pfizer’s descriptive clinical comparability analysis? Q37
Question 38: What was the regulatory basis for issuing Pfizer’s EUA in the absence of this analysis? Q38
Question 39: What actions did FDA take when Pfizer failed to submit its descriptive clinical comparability analysis by the specified date? Q39
Question 40: What was the regulatory basis for re-issuing Pfizer’s EUA with its various amendments including those involving booster shots and new variant versions in the absence of this analysis? Q40
Question 41: Please confirm the information provided by MHRA in their FOI response 23/510 that this analysis was never conducted and submitted to FDA. Q41
Question 42: Please confirm the information provided by MHRA in their FOI response 23/510 that analysis was removed from the protocol in amendment 20 in September 2022. Q42
Question 43: What was the justification provided by Pfizer for not conducting or submitting this analysis? Please confirm that all or part of this justification is similar to that provided by MHRA in their FOI response 23/510 that this was “due to the extensive usage of vaccines manufactured via “Process 2.” Q43
Question 44: Comparing and contrasting with Question 32 and noting FDA’s 1998 (19), 2019 (20), and 2023 (21) guidance documents regarding evidentiary standards for clinical data, what is the regulatory basis for authorizing or approving a vaccine based on only one clinical study of DP made by a process that differs with DP currently used and made by a process for which there is no “substantial evidence” of clinical comparability “consisting of adequate and well-controlled investigations.” Q44
Question 45: If FDA is relying on “extensive usage” in a manner apparently similar to MHRA, is this intended to constitute Real World Evidence (RWE) that can support approvals under some circumstances only described in FDA’s September 19 guidance? (21) Has this RWE been subject to the appropriate controls described in a guidance only recently (Aug 30, 2023)? (41) Q45
Question 46: Was Process 2 DP used in any of Pfizer’s other trials or sub-trials? If so, which? Q46
3.3. EMA concerns regarding the Pfizer process and the process change
Although, as mentioned above, EMA in their February 2021 assessment decided that, given the emergency situation, a number of uncertainties would be addressed after issuing a conditional authorization. (30) While EMA determined that comparability between the two processes had been adequately demonstrated, several matters remained sufficiently concerning to impose “Specific Obligations” in addition to a number of “ Recommendations.” Although some matters were later considered resolved by EMA, with some detail being published by Pfizer, (42) this is by no means settled science and their reexamination is warranted in light of recent findings concerning plasmid-derived DNA (section 5 ), frameshift proteins (section 14 ) and lipid nanoparticles, lipid adducts (section 8 ) discussed below.
Pertinent to this discussion of DNA and RNA, EMA had questions, Specific Obligations ( SO ) or Recommendations ( REC ) (p36/140) related to:
· SO1: the poly(A) tail pattern (p19/140)
· SO1: the 5’ cap (p18/140)
· SO2, REC8: mRNA integrity and dsRNA (pp21, 17/140)
· SO1: differences in the pattern and identity of RNA species revealed by electropherogram (p18/140)
Although EMA considered fragmented and truncated RNA unlikely to result in expressed proteins, EMA observed that the lack of experimental data did not permit a definitive conclusion. EMA noted (p137/140): “The high levels of these impurities reflect the instability of RNA resulting in generation of RNA fragments both in the transcription step and thereafter.”
· SO1: the identity and molecular weights of bands of expressed proteins observed in Western Blots; accounting for glycosylation as a source of discrepancies in molecular weight estimates from Western Blots. (p19/140)
· SO3: confirm the consistency of the finished product (commercial scale) manufacturing process (p36/140)
· REC3: Implementation of analytical methods for release testing of enzymes used in the manufacturing process. (p16/140)
· REC4 : Reassessment of the specification for linear DNA template purity and impurities.
· REC7 : The robustness of the DNase digestion step for the control of residual DNA impurities in DS, EMA noted the ongoing studies on this topic. (p17/140)
· REC10 : Suitable assay for biological characterization of protein expression for the active substance (p20/140)
Question 47: Did FDA express any concern to Pfizer about any of the process-related issues identified above, including the poly(A) tail pattern. the 5’ cap, mRNA integrity, dsRNA, the pattern and identity of RNA and truncated or fragmented RNA, and the identity and molecular weights of proteins expressed after modRNA transfection? How were these concerns resolved? What was the timeline of this process from FDA’s first awareness, to FDA’s expression of concern or questions, to Pfizer’s response and to resolution? Q47
Question 48: Were there concerns similar to those listed in Question 47 regarding the Moderna product? Please describe. Q48
Question 49: Was FDA aware of the concerns expressed by EMA or other regulatory agencies on the subjects discussed in Question 47 and the actions they took to address them? When? Was there any consultation or coordination between agencies? Q49
3.4. EMA concerns regarding residual DNA
Dr. Marks’ letter makes two statements concerning the removal of residual DNA:
· “As part of the purification process during production, the mRNA is treated with DNAse to digest residual DNA.”
· “The treatment of the products with DNAase also fragments any residual DNA template that might be present after other manufacturing steps.”
No doubt these statements were intended to allay concerns about residual DNA. Dr. Marks’ letter fails to acknowledge that concerns persisted about residual DNA and the DNase digestion step, well past Pfizer’s 2020 EUA and 2021 BLA as revealed in EMA documents.
Reports relating to most of EMA’s Specific Obligations (SO) were to be submitted by March 2021. SO3 as well as three (3, 4, 7) of the recommendations from EMA’s February 2021 assessment (30) listed above related in whole or part to various aspects of the DNA template or residual DNA. Two documents released recently pursuant to the European public access regulations (ASK-148075, October 25, 2023) suggest that residual DNA had remained an ongoing issue even to June 2022.
Below are two screen shots from the first of these redacted documents, “CHMP Assessment Report for the Post-Authorisation Measure REC 027, Comirnaty” submitted March 2021 with conclusions adopted May 2021 (43) The redactions are original to the EMA documents as disclosed.

In the context of the third sentence of this first screenshot that begins “As a mitigation approach…” and discusses the robustness of the DNase digestion step , the second word of the paragraph, which, following the word “An,” must begin with a vowel, appears to be ”excess,” “elevation,” or similar.
This is consistent with an unredacted document dated November 25 2020 “ Rapporteur’s Rolling Review assessment report Overview and list of questions. Procedure No. EMEA/H/C/005735/RR.” that states:
"Residual DNA template is present at higher level in PPQ3 [8] batch (211 ng DNA / mg RNA) than in PPQ1 and PQ2 batches (10 and 23 ng/mg); the robustness of DNase I digestion step should be further investigated." (p16/78)
and
“No validation data are available to confirm consistent removal of impurities, which is not acceptable. In addition, residual DNA template is present at higher level in PPQ3 batch (211 ng DNA / mg RNA) than in PPQ1 and PPQ2 batches (10 and 23 ng/mg) which does not confirm the robustness of DNase I digestion.” (p58/78)
This document [9] is among those leaked in December 2020 to several journalists (44) and the BMJ. (45) Given their use by the BMJ, the passage of three years in which the authenticity of the documents could have been challenged, as well as consistency with other documents of known provenance, this document is likely authentic.
A second screen shot refers to Recommendation 3 ( release testing of enzymes used in the manufacturing process) directly and as a “request” with time frame of 2Q2021.

The words “robustness of the DNase digestion step” is a reference to Recommendation 7. As in the earlier screen shot, the first redaction appears to signal “excess” or similar, or perhaps “variable” or similar. The second redaction appears to be referring to some quality deficiency in the DNase leading to concerning residual DNA levels.
The second document released pursuant to European public access regulations (ASK-148075) is titled “CHMP Type IB variation report, Comirnaty.” (46) The document relates to an application by Pfizer made on January 24 2022 to, inter alia, implement test procedures to qualify DNase used in manufacturing, stemming from Recommendations 3 and 7, to improve the robustness of the DNase digestion step, and therefore result in consistently and acceptably lower levels of residual DNA.
As shown in the screenshots below, the goal of each recommendation was only partly fulfilled. A revised submission was planned for June 2022.


What is most concerning is that some 18 months (at least) after the Pfizer EUA was issued, and about nine months after the COMINARTY BLA was approved, issues with residual DNA still remained.
Question 50: Did FDA express any concern to Pfizer about any issue related to residual DNA such as the robustness of the DNase digestion step. How were these concerns resolved? What was the timeline of this process from FDA’s first awareness, to FDA’s expression of concern or questions, to Pfizer’s response and to resolution? Q50
Question 51: How is the continuing concern well into at least 2022 about residual DNA consistent with the issuance of COMIRNATY’s BLA in August 2021 and the authorization of children’s doses in October 2021? Q51
Question 52: Were there concerns similar to those listed in Question 50 regarding the Moderna product? Please describe. Q52
Question 53: Was FDA aware of the concerns expressed by EMA or other regulatory agencies on the subjects discussed in Question 50 and the actions they took to address them? When? Was there any consultation or coordination between agencies? Q53
3.5. What is “GMP-like”?
Dr. Marks makes two statements that reference the manufacturing process:
· “We would like to make clear that based on a thorough assessment of the entire manufacturing process, FDA is confident in the quality, safety, and effectiveness of the COVID-19 vaccines.”
· “FDA takes its responsibility for ensuring the safety, effectiveness and manufacturing quality of all vaccines licensed in the U.S., including the mRNA COVID-19 vaccines, very seriously.”
A (June 21 2023) webinar hosted by the USP entitled “ Quality Considerations for Plasmid DNA as a Starting Material for Cell and Gene Therapies” included a presentation by Lawrence Thompson, Associate Research Fellow, Pfizer, and a member of the USP Expert Panel to develop a USP general chapter on “manufacturing and testing of plasmid DNA used as a starting material in the manufacturing of CGT [10] / mRNA products.”
Mr. Thompson discussed the quality of plasmids used in manufacture: [11]
“Then around 2015, we started moving into the gene therapy area, specifically aav gene therapy. And we started working with suppliers who had materials, and they were calling them GMP-like materials, all right, this is sort of a brand new thing that started to pop up. It's not research grade, it's not GMP. What does GMP like even mean? We could talk about that a little more in the discussion section where they have some controls and you need to have some oversight of that. What's the quality of that material? You're going to take a lot of time with supplier knowledge and scientific justification to sort of figure that out. This is when we were working with supplier. Then we started moving internally and making our own stuff, and we had a mix of these different materials. Again, there was a lot of phase-appropriate scientific justification, a lot of discussions about that for this, again, for the gene therapy. Then in 2020, everyone's well aware, we were able to pivot our pipeline from not just gene therapy to mRNA. And there was a whole separate new group of questions that we had to ask ourselves.”
Mr. Thompson revealed that around 2015 suppliers were providing “GMP-like” grade plasmid. From the context of the transcript and the accompanying slide below, it is clear that in 2020 for mRNA manufacturing “GMP-like” plasmid was being used.
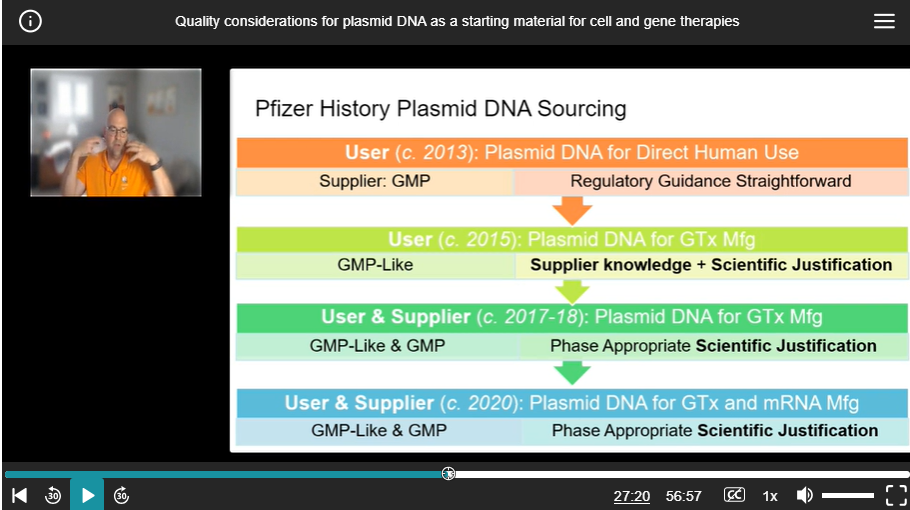
Figure 3 : Reference to “GMP-like” in presentation by Pfizer’s Mr. Thompson
Although the presentation and its slide discuss “ phase appropriate scientific justification,” it is unclear what this means.
Question 54: Is FDA aware that “GMP-like” may have been used in the manufacture of Pfizer mRNA? Q54
Question 55: For which phases of Pfizer COVID-19 vaccine preclinical, clinical and post EUA use was “GMP-like” plasmid used? Q55
Question 56: What was the source of Pfizer’s plasmid and its GMP provenance (i.e. GMP, or GMP-like)? Was this obtained from Pfizer’s Gene Therapy Division at the large-scale pDNA manufacturing facility in Chesterfield, MO? Was FDA aware of the source of plasmid? Q56
Question 57: How does “GMP-like” plasmid differ from GMP-compliant plasmid? Q57
Question 58: Regarding the COVID-19 mRNA vaccines, for what other preclinical, clinical and post EUA purposes were processes or materials “GMP-like” rather than “GMP compliant? Were these instances of “GMP-like” a reflection of EUA regulations or FDA’s non-enforcement of GMP requirements? Q58
Question 59: Where there any instances of “GMP-like” processes or materials related to the development or manufacturing of the Moderna COVID-19 mRNA vaccine? Q59
3.6. Formulation and process changes after nonclinical studies
A number of major and minor changes are known to have been implemented following initial nonclinical studies that could affect a number of quality attributes of modRNA products discussed in this document.
Question 60: For the Pfizer product, which process was used to make the drug product tested in the non-clinical pharmacology and toxicology studies described in the Summary Basis for Regulatory Action. (47) Was test article taken from clinical or commercial scale production material, or from especially conducted non-clinical runs? Q60
Question 61: For the Pfizer product, which non-clinical studies were performed with the V8 version and which with the V9 version? Please confirm that the type made by both Processes 1 and 2 was the V9 type. Q61
Question 62: For both the Pfizer and Moderna products, please summarize and tabulate differences in the composition of Drug Product used in non-clinical, clinical, and post-authorization COVID-10 vaccines, paying particular attention to the modRNA ORF sequence, sequence of non-coding regions, extent and type of nucleoside medication, pattern of codon optimization, LNP and buffer composition. Please indicate the reason for each change and what analytical, non-clinical or clinical comparability studies were performed to qualify these changes. Q62
Question 63: For both the Pfizer and Moderna products, please summarize and tabulate all manufacturing changes from the formulation and process used to produce non-clinical test material to currently produced vaccine that may have changed the amount, type of size distribution of DNA or RNA in the final DP, the amount and type of impurities, as well as critical quality attributes and properties of the LNPs. Please indicate the reason for each change and what analytical, non-clinical or clinical comparability studies were performed to qualify these changes. Q63
The relevance and reliance on supportive studies involving related modRNA products with similar, but not identical compositions will be discussed in sections 11.4.2 and 12.3.1 .
4. Why are the possible harms from residual DNA?
4.1. Prior downgrading of integration risk for residual DNA
Dr. Marks’ non-concern for an integration risk of residual DNA perhaps reflects comments in a 2021 version (48) of the 2007 WHO (49) document addressing DNA vaccines positing that “prior concerns about integration, autoimmunity and immunopathology have not been borne out” and speculating that “the observed reactogenicity appears to relate more to the delivery method than to the DNA vaccine itself, most notably in the case of electroporation or particle-mediated bombardment.” (p14/54)
The downgrading of integration risk has evolved over many years. The 2013 WHO Recommendation document regarding cell substrates for biological products (50) provides an interesting historical perspective on risk assessment related to residual DNA. (p8/110)
In the 1970’s as a result of a clinical research need, interferon alpha (IFN-α) was produced in a tumor line whose cells contained the Epstein-Barr virus genome. With a concern that viral DNA could be transmitted in whole or part to patients, regulatory agencies in several countries authorized human clinical studies and eventually approved an IFN-α product . Contributing to those decisions and the risk-benefit consideration was the fact that the product was being used for therapeutic rather than prophylactic purposes, and that a validated purification process along with assays that could show that DNA was undetectable within the assay limits.
Nonetheless, the same document (50) acknowledged and reviewed the risk of residual DNA en passant (p22/110), citing several papers. (51-55) One particular paper cited, written in 1995 by Petricciani [12] and Horaud (56) is remarkable in that its authors appear to hold, simultaneously contradictory positions that approximate to those at the heart of our current discord.
On the one hand Petricciani and Horaud view DNA and its threats in biologic products in terms of “DNA dragons,” stating that “ many of us believe that we can now see the DNA dragons for what they are: real myths.”
On the other hand, this expression of “belief” appears tempered by its sandwiching between more factual descriptions of the state of science on this topic. This declaration of belief seems to have been precipitated by work bemoaningly cited as demonstrating that an inefficient extraction procedure grossly underestimated the amount of DNA in a biological sample, with the assertion, appropriately, that “ if tests for DNA are to be required, the results should be meaningful rather than fanciful. In other words, the numerical values reported should not be presented simply to provide comfort to regulatory authorities that risk has been reduced, but they should have scientific credibility and be biologically meaningful.”
Petricciani and Horaud concede that “Nevertheless, it also is clear that more needs to be done to achieve a final resolution of the DNA issue. Complex problems are not solved easily or quickly, especially when there is an emotional element overlaid onto scientific issues.” They call for an international group with broad regulatory, industrial, and academic representation to answer questions, including:
· “Is residual cellular DNA a practical and realistic risk, or simply a theoretical concern?
· What range of cellular DNA levels should be considered essentially risk-free?
· What levels of cellular DNA should be considered acceptable as an impurity in biological products?”
In the span of some 30 years and with advances in modRNA and lipid nanoparticle technology, these questions remain as relevant today as in 1995. Most concerning is that Petricciani and Horaud’s motivation in calling for an international group appears to reach a foregone conclusion (theirs) rather than conduct healthy scientific discourse or consider opposing arguments:
“The time is long overdue for sanity to return to the issue of residual cellular DNA in biological products, and for DNA to be treated as a simple impurity rather than as a monster. A conference such as the one proposed above would provide a mechanism to achieve that goal.”
Petricciani and Horaud may well have been correct in their assessment of risks associated with residual DNA resulting from the vaccine technology of the 1990s. But while technology has progressed with innovations in modRNA and LNPs over 30 years, we have not progressed in our ability to grapple with “[c] omplex problems [that] are not solved easily or quickly, especially when there is an emotional element overlaid onto scientific issues.” The time is indeed “long overdue for sanity to return to the issue of residual [cellular] DNA” and that FDA’s detailed answers to the detailed questions posed here, devoid of over-simplification, obfuscation, and thinly veiled accusations of misinformation, will “ provide a mechanism to achieve that goal.”
4.2. Risk of chromosomal integration: nuclear access of DNA
Dr. Marks’ letter makes two statements concerning the plausibility of chromosomal integration of residual DNA, arguing that since access to the nucleus is unlikely, integration is even less likely:
“On first principle, it is quite implausible that the residual small DNA fragments located in the cytosol could find their way into the nucleus through the nuclear membrane present in intact cells and then be incorporated into chromosomal DNA.” (his footnote 2 citing (57) )
“Regarding concern for possible integration of the residual DNA fragments into reproductive cells: Please see the response to the first question above regarding the implausibility that the minute amounts of small DNA fragments present could find their way into the nucleus of these cells.”
These assertions of the implausibility of genomic insertion by residual DNA rely solely on the one reference cite by Dr. Marks as footnote 2 (57) , a chapter in a textbook by Cooper, “The Cell: A Molecular Approach.” The author’s preface reads (emphasis added):
“The goals and distinguishing features of The Cell, however, remain unchanged from the first edition. The Cell continues to be a basic text that provides an accessible introduction for undergraduate or medical students who are taking a first course on cell and molecular biology.”
The chapter cited describes the fundamental properties of the nuclear membrane, whose properties are the basis for the assertion as to im plausibility of insertion. It does not discuss the plausibility of nuclear entry or genomic insertion of residual DNA, nor would such a basic introductory undergraduate text be expected to do so. [13] However, from the same basic text book cite by Dr. Marks, another chapter entitled “The Nucleus during Mitosis” (58) renders meaningless the suggestion of “first principle” implausibility of DNA entry by describing how the nuclear membrane breaks down during mitosis, thus plausibly providing access of residual DNA to chromosomal DNA. The chapter states:
“A unique feature of the nucleus is that it disassembles and re-forms each time most cells divide. At the beginning of mitosis, the chromosomes condense, the nucleolus disappears, and the nuclear envelope breaks down , resulting in the release of most of the contents of the nucleus into the cytoplasm .”
Dr. Marks’ statements would in fact be relevant to yeasts, as Cooper elaborates: “ Some unicellular eukaryotes (e.g., yeasts) undergo so-called closed mitosis , in which the nuclear envelope remains intact.” However, as Boettcher and Barral (59) confirm, human cells undergo open mitosis.
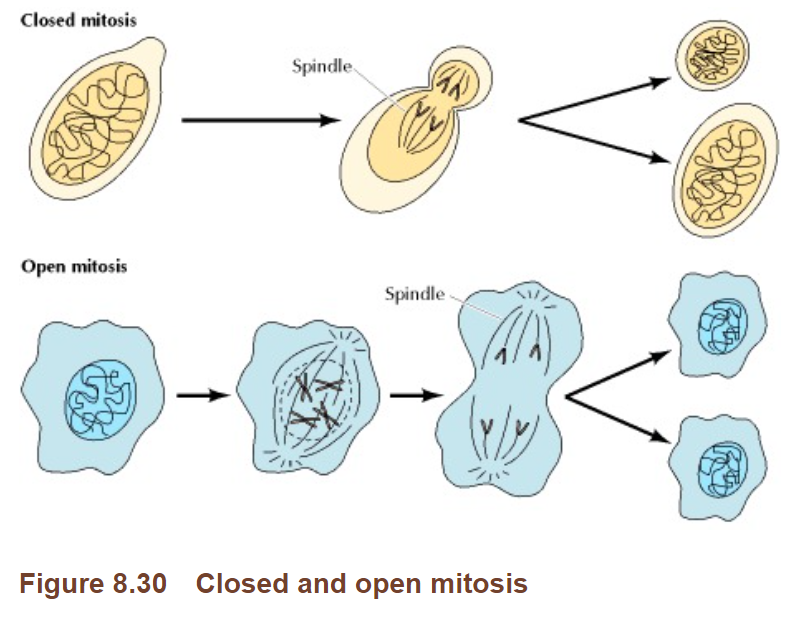
Figure 4 : Figure 8.30 from Cooper
Indeed, Faurez et al., (60) in discussing the biosafety of DNA vaccines and DNA entry into the nucleus, list incorporation of plasmid DNA into the nucleus “ during disassembly of the nuclear membrane during mitosis” as one mechanism, alongside nuclear import by simple or facilitated diffusion through nuclear pores.
Lechardeur et al. (61) review the diffusion of DNA fragments smaller than 250 bp into the nucleus via the Nuclear Pore Complex (NPC).
Question 64: In light of the above review of basic cell biology, and FDA’s “first principle” premise of nuclear membrane inviolability, what studies have FDA conducted or solicited or will FDA be conducting or soliciting from Pfizer and Moderna regarding the intracellular kinetics of residual DNA? Q64
If true, the assertion that DNA could not plausibly even enter the nucleus lacks substance and would render any guidelines concerning residual DNA levels pointless. This assertion is contradicted by the literature showing spontaneous integration after transfection (e.g. (62) ) and self-evident statements about DNA made in several documents authored by FDA, WHO, Moderna or BioNtech (emphasis added):
· FDA 2010 guidance: Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications (63) (cited in Marks footnote 4)
“Residual DNA might be a risk to your final product because of oncogenic and/or infectivity potential. There are several potential mechanisms by which residual DNA could be oncogenic , including the integration and expression of encoded oncogenes or insertional mutagenesis following DNA integration.” (p40/50)
· FDA 2007 guidance: “Considerations for Plasmid DNA Vaccines for Infectious Disease Indications. Guidance for Industry .” (64) [14]
“Theoretical concerns regarding DNA integration include the risk of tumorigenisis if insertion reduces the activity of a tumor suppressor or increases the activity of an oncogene. In addition, DNA integration may result in chromosomal instability through the induction of chromosomal breaks or rearrangements .” (p9/13)
· WHO 2007 Technical Report Series No 941, Annex 1. Guidelines for assuring the quality and nonclinical safety evaluation of DNA vaccines. (49)
“The injected DNA taken up by cells of the host may integrate into the host's chromosomes and cause an insertional mutagenic event.” (p17/25)
“It is known that DNA taken up by mammalian cells in culture can integrate into the cellular genetic material and be faithfully maintained during replication. This is the basis of the production of some recombinant therapeutic proteins.” (p18/25)
· Sheng-Fowler et al. [FDA staff] Issues associated with residual cell-substrate DNA in viral vaccines. (65)
“The presence of some residual cellular DNA derived from the production-cell substrate in viral vaccines is inevitable. Whether this DNA represents a safety concern, particularly if the cell substrate is derived from a tumor or is tumorigenic, is unknown. DNA has two biological activities that need to be considered. First, DNA can be oncogenic; second, DNA can be infectious/”
· US Patent Application assigned to Moderna US 2019 / 0240317 A1
“The direct injection of genetically engineered DNA (e.g. naked plasmid DNA) into a living host results in a small number of its cells directly producing an antigen, resulting in a protective immunological response. With this technique, however, comes potential problems, including the possibility of insertional mutagenesis , which could lead to the activation of oncogenes or the inhibition of tumor suppressor genes .”
· US Patent assigned to Moderna 2018 US 10,077,439 B2
“The DNA template used in the mRNA manufacturing process must be removed to ensure the efficacy of therapeutics and safety, because residual DNA in drug products may induce activation of the innate response and has the potential to be oncogenic in patient populations. Regulatory guidelines may also require the quantification, control, and removal of the DNA template in RNA products. Currently available or reported methods do not address this deficiency.”
· Sahin [founder BioNTech], Karikó [Nobel Prize 2023] and Türeci “mRNA-based therapeutics -developing a new class of drugs” (66)
“In addition, IVT mRNA-based therapeutics, unlike plasmid DNA and viral vectors, do not integrate into the genome and therefore do not pose the risk of insertional mutagenesis.”
· Dr. Robert Langer [co-founder Moderna] co-author in Reichmuth et al . “mRNA vaccine delivery using lipid nanoparticles” (67)
“mRNA, with the cytosol as its target, is easier to deliver and much safer than DNA, because the mRNA in the cytosol does not interact with the genome in the nucleus and is only transiently expressed. “
There are three other mechanisms whereby DNA might access the nucleus:
· Dalby et al (68) discuss evidence that Lipofectamine 2000, a commonly used laboratory transfectant, may promote “may promote penetration of DNA through intact nuclear envelopes” in some cell types.
· The work of Dr. David Dean at the University of Rochester [15] that focusses on n uclear t argeting of p lasmids and p rotein-DNA c omplexes has shown that t he SV40 enhancer/ promoter/ori sequence facilitates the localization of DNA in the nucleus, “especially in non-dividing cells.” (69, 70) I am informed that this is the same sequence as was found in the Pfizer vaccine (see 7.2 )
· Via a LINE-1 protein product and ribonucleoprotein pathway ( 4.3 ).
Question 65: In light of the above attestations as to the risks of insertional mutagenesis, would FDA revise Dr. Marks’ earlier statement concerning the plausibility of risk of chromosomal integration of residual DNA? Q65
4.3. Additive risks of integration by residual DNA and by DNA reverse transcribed from modRNA
Any risk of integration from residual DNA must be added to the risk associated with DNA produced by reverse transcription of vaccinal modRNA. In a 2022 interview posted on FDA’s Youtube channel, [16] Dr. Marks was asked to comment on “ the thought that vaccines can, particularly with the mRNA, alter one's DNA.” He gave three reasons to support his statement that “there's no way they can alter your DNA”:
· the mRNA is destroyed
· the RNA can't get into the nucleus, the nucleus has a one-way valve
· our cells do not have the enzyme necessary to get RNA back into DNA
These reasons mirror those given in the 2021 WHO guidelines on mRNA vaccines (71) (p47/66) to justify why: “nonclinical studies do not need to be performed to specifically address integration or genetic risks as these are considered to be theoretical issues for mRNA vaccines.” (p48/66)
As will be discussed, none of these arguments hold water, with evidence that these issues are more than “theoretical,” thereby raising concern and the need for studies “ to specifically address integration or genetic risks.”
4.3.1. “the mRNA is destroyed”
The notion is more fully expressed in the 2021 WHO guidelines on mRNA vaccines (71) : “the vaccine mRNA degrades within a relatively short time once taken up by the body’s cells, as does the cell’s own mRNA. During that entire time, the mRNA vaccine is expected to remain in the cytoplasm, where it will be translated and then degraded by normal cellular mechanisms.” (p48/66)
Although Pfizer did not appear to conduct RNA or protein metabolism studies, a number of reports from animal or human studies have now emerged that detect vaccinal modRNA or spike protein in various tissues weeks or months after vaccination (72-77) , see review in (78) . Based on all of these studies, it is clear that the premise that “mRNA is destroyed” to support the argument that “there's no way [the mRNA vaccines] can alter your DNA” is untenable and at best inconclusive.
Further discussion on the need for biodistribution studies including cellular kinetics of both vaccinal RNA and spike protein, is given in section 0 .
4.3.2. “the RNA can't get into the nucleus, the nucleus has a one-way valve”
Just as nuclear membrane disassembly during mitosis provides access of residual DNA to chromosomal DNA ( 4.2 ), it could apply also to RNA.
An additional potential mechanism is suggested by Sattar whose colleagues include those from NIAID in a work entitled “Nuclear translocation of spike mRNA and protein is a novel pathogenic feature of SARS-CoV-2.” (79) They described how the SARS-Cov-2 spike protein (but not that of other coronaviruses) contains a nuclear localization signal that enables them to translocate into the nucleus in virus-infected cells. Further, they found that spike mRNA also translocates into the nucleus, colocalizing with, and aided by spike protein.
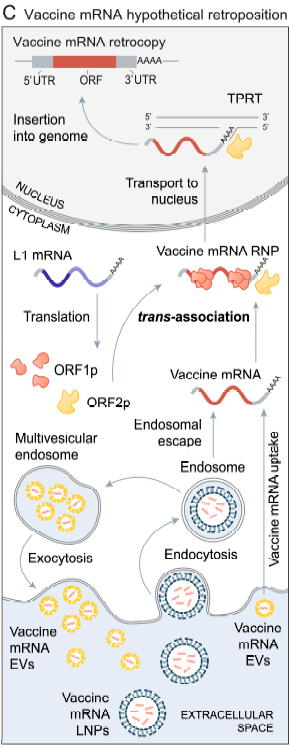
The premise of a “one-way valve” i s subject to challenge for another reason. Citing a rich literature, Domazet-Lošo (80) has reviewed the possibility that mRNA can retropose (undergo r everse transcription to DNA with subsequent insertional mutagenesis, see below) mediated by the protein products of LINE-1. These genes are part of the family of Long Interspersed Nuclear Elements and are considered “Transposable Elements” (TE) because they can transpose from one genomic region to another, modifying its structure and acquiring the name “jumping genes.” (81)
Domazet-Lošo proposes the mechanism illustrated below in which some of the protein products of LINE-1 form ribonucleoprotein complexes with vaccinal mRNA, possibly facilitating its entry to the nucleus, where it can undergo retroposition.
|
|
Figure 5 : Figure 1C and legend from Domazet-Lošo (80) “Hypothetical L1-mediated retroposition of vaccine mRNA. Vaccine mRNA formulated in lipid nanoparticles (LNPs) enter the cell via endocytosis. A fraction of the vaccine mRNA enters the cytosol via endosomal escape, while the rest of the vaccine mRNA undergoes degradation in endosomes or is repackaged in multivesicular endosomes into extracellular vesicles (EVs) and secreted back into the extracellular space. The neighboring or distant cells can uptake vaccine mRNA from these EVs. L1 proteins (ORF1p and ORF2p) interact with vaccine mRNA via a process termed trans-association to form a vaccine mRNA ribonucleoprotein particle (vaccine mRNA RNP). Like L1 and parental gene RNPs, a vaccine mRNA RNP enters the nucleus where the vaccine mRNA, through TPRT, is reverse-transcribed and integrated into the genome. The poly-A tail of vaccine mRNA plays a crucial role in this process.” (in text citations removed) |
Question 66: As with conventional pharmacokinetics (PK) (see 0Error! Reference source not found.), a full understanding of the cellular kinetics of any drug is essential to understand its pharmacology and toxicology and is not an academic nicety. What studies will FDA be conducting on its own, or soliciting from Pfizer or Moderna, regarding the intracellular kinetics of modRNA? Q66
4.3.3. “our cells do not have the enzyme necessary to get RNA back into DNA”
The 2021 WHO guideline on mRNA vaccines (71) (p47/66) expresses this premise as “The only known mechanism by which RNA can integrate into the host genome requires the presence of a complex containing reverse transcriptase and integrase.”
This premise is untenable given the reverse transcriptase activity of the protein products of endogenous LINE-1. In his review of retroposition, Domazet-Lošo (80) noted that in addition to the formation of ribonucleoproteins that may facilitate vaccinal modRNA entry into the nucleus, LINE-1 protein products display reverse transcriptase and endonuclease activities. (80) Based on the abundance of LINE-1, and reviewing a number of pertinent factors that could increase retroposition (N-1 methyl pseudouridylation, use of LNP, improved stability, half-life and translational efficiency), Domazet-Lošo voices an “urgent[ly] need [for] experimental studies that would rigorously test for the potential retroposition of vaccine mRNAs. At present, the insertional mutagenesis safety of mRNA-based vaccines should be considered unresolved. ”
Further evidence (see review in (82) ) for the ability of endogenous LINE-1 to effect reverse transcription, and possible insertion comes from the work led by Dr. Rudolf Jaenisch from the Whitehead Institute, MIT and the National Cancer Institute, and funded partly by NIH. (Zhang et al., (83) ) Noting in their introduction, detection of non-retroviral RNA viral sequences into the genomes of many vertebrates, Zhang et al. studied cell lines and patient-derived tissues and found evidence for reverse-transcription of SARS-CoV-2 viral RNA and genomic integration by a LINE-1 mechanism. This could result, possibly by template switching, in the production of human-viral mRNA chimeric transcripts and raising the prospect of novel proteins.
In a subsequent work, the same group reported genomic integration of SARS-CoV-2 mRNA mediated by LINE-1 associated reverse transcription into human cell lines infected with virus, (84) again controverting the premise that “ our cells do not have the enzyme necessary to get RNA back into DNA.” This was not observed for cells transfected with viral RNA. The authors point out a number of limitations to extrapolating their finding to conclude that this would not occur with modRNA vaccines. Firstly, these studies were performed in cell lines and not animals or humans. Secondly, their study achieved transfection using lipofectamine rather than LNPs. Thirdly, the RNA sequence in the study was nucleocapsid and not spike protein. Another important difference was in the sequences of the untranslated regions, the use of N1-methyl pseudouridine and the use of codon of optimization. The findings of Zhang et al. have been challenged, and discussed further by two of the works’ coauthors. (85)
Many of these limitations were absent in the paper by Alden et al., (86) who reported that BNT162b2 mRNA was reverse transcribed into DNA within six hours after human liver cell line cells were exposed to vaccine in vitro . This finding dispels another argument made by the 2021 WHO guideline on mRNA vaccines (71) to dismiss the need for integration studies: “Further, the design of candidate mRNA vaccines should be considered so that they do not include specific RNA-binding sites for primers required for the reverse transcriptase to initiate transcription.” (p47/66)
Alden et al. also reported that the vaccine increased distribution of LINE-1 protein in the nucleus. The effect may not be due to the primary components of the Pfizer vaccine, but rather to double-stranded RNA (dsRNA), a process-related impurity, as Zhang et al., (84) suggested that dsRNA, known to induce innate immune responses, could induce endogenous LINE-1 expression. They further noted “ that therapeutic mRNAs should be designed to avoid unwanted dsRNA (secondary) structures.” Since dsRNA is a process-related impurity, variability between lots could contribute to variable vaccine reactions.
Another reason why the premise that “our cells do not have the enzyme necessary to get RNA back into DNA” is untenable because reverse transcriptase may be expressed by genes from Human endogenous retroviruses (HERVs). HERVS are ancient retroviral genomic integrations estimated to account for about 8% of the human genome and possibly involved in a number of diseases including cancer, autoimmune, and neurodegenerative disease. (87) HERVs related genes may be upregulated or activated by environmental factors such as viruses, including influenza A (88) and SARS-CoV-2. (89, 90) These findings have implications particularly in vaccinees that have also been infected with SARS-CoV-2.
Question 67: In light of the above evidence, what studies is FDA conducting in its own labs, is aware of being undertaken by other government agencies, or is soliciting from Pfizer of Moderna to characterize the reverse transcription of vaccinal modRNA to DNA? Q67
Question 68: In light of the above evidence, what assessments have FDA conducted, solicited from Pfizer of Moderna or received from elsewhere to characterize the risks of reverse transcription of vaccinal modRNA to DNA? Q68
4.4. The need for integration studies
As detailed, there is sufficient reason to dismiss the non-concern for the need for integration studies, both for residual DNA ( 4.2 ) and potentially reverse transcribed RNA ( 4.3 ).
The WHO 2007 guideline (49) for DNA vaccines is instructive. The document discusses (p18/25) the fractions of naked DNA entering cells, entering the nucleus, inserting into DNA, inserting at a key location, leading to a low probability of tumorigenesis, borne out, at least partly, in some studies. The document continues to discuss circumstances where integration studies might be required:
:
“ However, alternative formulations or administration devices such as co-inoculation of a plasmid encoding a growth promoting factor, or electrostimulation, can lead to an increased potential for integration of plasmid DNA, and an investigation of the potential integration of the plasmid DNA in vivo into the host’s chromosomes should form part of the nonclinical safety testing of a DNA vaccine.
Integration studies may not be necessary for a plasmid DNA vaccine if prior information on a similar plasmid, with the same mode of administration already exists. There would be a need to reassess integration if there was a significant change in the method of delivery , especially any change potentially involving an increase in the capacity of plasmid DNA to enter the nucleus .” (emphasis added)
The use of LNPs certainly increases the capacity of DNA to enter the cell, (91-93) and therefore likely also the nucleus. Accordingly, integration studies would appear to be called for.
Question 69: What studies have FDA requested from Pfizer or Moderna to determine whether genomic insertion may occur with residual DNA or from reverse transcribed vaccinal modRNA after modRNA vaccine administration? Q69
The WHO 2007 guideline (49) cites two papers (94, 95) as examples of how an association of plasmid DNA with genomic DNA may be assessed.
Question 70: What in vitro or in vivo models does FDA consider suitable to assess genomic integration of residual DNA, after appropriate validation? Q70
Noted are a series of elegant attempts conducted in FDA’s own laboratories led by Dr. Keith Peden to develop an animal model capable of assessing the oncogenic activity of DNA. (Sheng-Fowler et al (96-98) ) Variously, tumors could be induced in athymic, immune-defective or transgenic mice, animals, newborn hamsters and rats after administration of oncogene-laden plasmid, particularly plasmid dually expressing H- ras and c -myc oncogenes. Sheng-Fowler et al. could induce tumor formation in small populations (n = 13 - 110) of mice with doses, in the case of linear plasmid, as low as 800 pg (97) or 300 pg (98) They were able to detect DNA integration in cells derived from induced tumors as well as expression of oncogene protein products. However, extracted DNA was unable to induce oncogenesis. The authors concluded that “ available in vivo models are not sensitive enough to detect the oncogenicity of cellular DNA.” (98)
The challenge of model development is readily appreciated, with even greater challenges instore to establish clinical relevance, even if successful in the lab. Nonetheless, further model refinements may bear fruit, perhaps if not as a model for oncogenicity, but as one for integration. One paper (96) reported that transfection facilitators were unable the efficiency of tumor induction, although these studies appear highly limited.
Question 71: What studies have FDA conducted, or will conduct to determine whether genomic insertion may occur with residual DNA or from reverse transcribed vaccinal modRNA? Please provide details. Q71
Question 72: Have FDA conducted studies using the models described in or adapted from Sheng-Fowler et al(96-98) to assess integration or oncogenesis after administration of the oncogene expression plasmids within the same or similar LNPs used in the Pfizer or Moderna COVID-19 vaccines? Please provide details. Q72
Question 73: Have FDA conducted studies using the models described by or adapted from Sheng-Fowler et al(96-98) to assess integration or oncogenesis after co-administration of the oncogene expression plasmids and sequence elements from the plasmid vectors used for modRNA COVID-19 vaccine production? Please provide details. Q73
Question 74: Have FDA conducted studies using the models described by or adapted from Sheng-Fowler et al(96-98) to assess integration or oncogenesis after co-administration of the oncogene expression plasmids and sequence elements from the plasmid vectors used for modRNA COVID-19 vaccine production within same or similar LNPs used I the COVID-19 vaccines? Please provide details. Q74
4.5. Risk of episomal / extrachromosomal expression
Insertional mutagenesis is not a prerequisite for expression of DNA introduced into cells. Indeed, this principle has been used to develop gene therapies by using episomal / extrachromosomal DNA eukaryotic vectors, as plasmids. (99) DNA produced by LINE-1 mediated reverse transcription from RNA, or RNA within sperm can be transferred to oocytes at fertilization and propagated during embryo formation. These sequences are extrachromosomal and can be transcribed to induce novel phenotypic traits. (100)
The risk of extrachromosomal expression of DNA is acknowledged by Moderna in its US Patent Application US 2013 / 0259924 A1:
“ Alternatively, the heterologous deoxyribonucleic acid (DNA) introduced into a cell can be inherited by daughter cells (whether or not the heterologous DNA has integrated into the chromosome) or by offspring.”
The SV40 promoter apparently of the same sequence as was found in the Pfizer pro-vaccine has been shown in cell culture to enhance expression of episomal DNA transgenes, without needing insertional mutagenesis" (101)
Question 75: What studies have FDA conducted, will conduct or have solicited from Pfizer or Moderna, to determine whether extrachromosomal expression or transmission of residual DNA occurs, and to determine the attendant risks, if detected? Q75
4.6. Non-integrating mechanisms of DNA toxicology
Residual DNA may have toxic effects unrelated to genomic insertion.
Immunomodulatory activity
A paper by FDA staff Sheng-Fowler et al, (65) noted “A nother biological activity of DNA that does not require gene expression is the immunomodulatory activity of DNA itself [9,10]. While this immunomodulatory activity can be measured in vivo, the amounts of DNA required for this effect are not typically present in vaccines .”
dsDNA in plasmid DNA vaccines may mediate adjuvanticity contributing to immunogenicity via a TANK-binding kinase 1 pathway (102) or via toll-like receptor 9 (TLR9). A recently approved hepatitis B vaccine (Heplisav B) uses bacterial CpG motifs as an adjuvant. (103)
Effect on pattern receptors and innate immunity
DNA can stimulate the innate immune response ( (104) , cited in (48) ) This property is one reason why its removal is said to be important in the production of mRNA. ( Moderna patent US 10,077,439 B2) The effect of dsDNA as an activator of innate immunity via intracellular pattern recognition receptors has been described regarding prothrombotic effects vascular endothelium . (105) This is particularly concerning given the recent disclosure by VSD lead Dr. Nicola Klein at the September 12 2023 ACIP meeting of signals for acute myocardial infarction and venous thromboembolism. (106) as well as signals published by FDA staff for pulmonary embolism, acute myocardial infarction, and disseminated intravascular coagulation. (see also Error! Reference source not found. ) An extensive review has been published of DNA’s action as a damage-associated molecular pattern (DAMP) able to drive inflammation via a number of pattern recognition receptors (PRR) including cGAS, AIM2, NOD, LRR, NLRP3, RAGE, TLR9. (107)
An association has been reported between ischemic stroke and the cGAS-STING pattern-receptor inflammatory pathway that can be triggered by pathogen, bacterial or viral dsDNA. (108) Two recent preprints whose authors included FDA scientists (109) or VSD lead Dr. Nicola Klein (110) found possible associations between non-hemorrhagic stroke and COVID-19 vaccination (see also Error! Reference source not found. ).
Autoimmune reactions
WHO 2007 guidelines (49) in discussing the risks of injecting plasmid DNA includes the risk that: “Antibodies may be formed against the injected DNA itself and these may contribute towards undesired autoimmune reactions.” (p17/25) Further, it states that sensitive ELISA analysis form human and animal models “ have shown that repeated DNA vaccination can stimulate a ≤ 5-fold increase in anti-DNA auto-antibody levels.” [17]
The corresponding FDA document (64) [p8/13] expands the scope of this concern to an immune response against cells expressing the target antigen, but downplays the possibility of both based on preclinical data available at the time. Further, this FDA 2007 guidance “ no longer recommend that preclinical studies be performed to specifically assess whether vaccination causes autoimmune disease,” instead “recommend that the general welfare of animals in preclinical immunogenicity and toxicity studies continue to be carefully monitored.” The guidance nonetheless noted that “ Yet the possibility persists that DNA vaccines might idiosyncratically cause or worsen organ-specific autoimmunity by encoding antigens (including cryptic antigens [18] ) that cross-react with self.”
Question 76: Given that these guidelines did not contemplate the highly efficient transfection of nucleic acid by LNPs (see 6.2), please provide a justification as to why FDA’s original (pre-2007) recommendation to conduct preclinical studies to assess vaccine-induced autoimmune disease should not be reinstated? Q76
Question 77: Has FDA conducted or solicited from Pfizer or Moderna a risk assessment related to vaccine -associated autoimmune disease? Q77
Question 78: Has FDA communicated with other US government agencies such as NIH or CDC about the risk of modRNA vaccine -associated autoimmune disease? Q78
Question 79: Is FDA aware of risk assessments or studies performed by other US government agencies such as NIH or CDC related to the risk of modRNA vaccine -associated autoimmune disease? What is the nature of this work? Q79
Question 80: What studies or risk assessments has FDA conducted or will conduct, has solicited, or will solicit from Pfizer or Moderna, to determine the contribution of non-integrating mechanisms of toxicity of DNA to the overall safety profile of the modRNA vaccines? Q80
Question 81: What lessons regarding DNA toxicity can learned from the viral vector COVID-19 vaccines (Janssen, Astra-Zeneca) and applied to the toxicity of residual or reverse transcribed DNA associated with the modRNA vaccines? Q81
5. Experimental findings regarding residual DNA
Dr. Ladapo’s concerns were focused on the then recently preprinted findings related to residual DNA in Pfizer and Moderna modRNA vaccines. (2, 3) As Dr. Marks noted the existence of plasmid-derived residual DNA in not surprising as it is a known process related impurity. The main findings from this work were:
· The amounts of residual DNA per dose approached or exceeded the 10 ng/dose guidance limit, depending on the assay method used.
· Fragments larger than the 200 base pair length size described in guidelines (see 6.4 ) were found.
· In the case of the Pfizer product, three sequences apparently not disclosed to regulators, were found. These were 1) SV40 enhancer/promoter/ori; 2) SV40 poly(A) signal; 3) HSV-TK poly(A) signal.
· Intact sequences for the SV40 enhancer/promoter/ori and antibiotic resistance genes were found.
The authors acknowledged the obvious limitations to their work, and “urge that [their] work is replicated under forensic conditions.” (3) This work in various aspects has been confirmed by several laboratories (6-10) and, notably, by high school intern students working under FDA supervision. (11) Our own unpublished work has confirmed this with the Pfizer 2024-2025 JN.1 formula. Further, in numerous FOIA disclosures around the world, regulators have confirmed the presence of the SV40 sequence (including the Marks response (4) ) and the fact that Pfizer had chosen not to specifically disclose this regulatory sequence. One acknowledged limitation relates to interference by RNA in the measurement of DNA by Qubit fluorometry. Kammerer et al. (8) successfully reduced the fluorescence using RNAse, leaving a DNA signal corresponding to levels about the 10ng/ dose guideline (see below). There still remains the possibility that RNAse may not remove RNA that is hybridized to DNA. A number of factors are important in preparing samples for UV or fluorescence methods. Further, the absorption characteristics of N1 methylpseudouridinylated modRNA differ from those of uridine. (111)
These and other technical challenges in measurement of residual DNA are evident in industry forums, or efforts of the United States Pharmacopoeia (112, 113) who are only now beginning to develop to develop compendial standards and test methods. The absence of an agreed upon test method for residual DNA is perhaps reflected in EMA’s recently (March 2025) guidelines on the quality aspects of mRNA vaccines, which stated “Orthogonal test methods should be used to quantify and characterise residual DNA.” (114)
6. What are the guidelines concerning residual DNA?
Dr. Marks stated : “There are internationally agreed upon recommendations for the quantity of residual DNA present in all biological products, including the mRNA vaccines,” citing two documents:
· WHO 2007 (115) : “ WHO Study Group on Cell Substrates for Production of Biologicals”
This document is a meeting report for the WHO Study Group on Cell Substrates for Production of Biologicals that reviewed data that would form the basis for further recommendations. The members of WHO Study Group on Cell Substrates listed in the document included representatives from Prizer and Merck as well as FDA’s Dr. Lewis and Dr. Peden, both of whom addressed the meeting.
· FDA 2010 (63) : “ Guidance for Industry. Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications ”
In addition to these documents, several others are relevant.
· WHO -RNA 2021. (71) : “Evaluation of the quality, safety and efficacy of messenger RNA vaccines for 10 the prevention of infectious diseases: regulatory considerations. WHO/BS/2021.2402”
· FDA 2007 (64) : “Considerations for Plasmid DNA Vaccines for Infectious Disease Indications. Guidance for Industry .” As pointed out in Marks footnote 1, this guideline was written for DNA vaccines themselves, rather than for DNA as “ DNA as a contaminant in other vaccines.” However, this document, as we as others listed here, contain a number of principles instructive to this discussion.
· WHO. 2007 (49) : “Technical Report Series No 941, 2007. Annex 1. Guidelines for assuring the quality and nonclinical safety evaluation of DNA vaccines.”
· WHO 2013 (50) : “ Annex 3. Recommendations for the evaluation of animal cell cultures as substrates for the manufacture of biological medicinal products and for the characterization of cell banks Replacement of Annex 1 of WHO Technical Report Series, No. 878.”
· WHO – DNA 2021 (48) : “ WHO. Guidelines on the quality, safety and efficacy of plasmid DNA vaccines. Annex 2. TRS No 1028 Replacement of Annex 1 of WHO Technical Report Series, No. 941.”
6.1. Guidelines on amount of residual DNA
Both the FDA 2010 (63) (p40/50) and WHO 2007 (115) (p20/30) documents describe a limit for residual DNA at 10 ng/dose for parenteral administration.
Dr. Marks stated: “The specification for the COVID-19 mRNA vaccines for residual DNA following DNAse treatment results in the presence of DNA fragments at a quantity that is less than three orders of magnitude lower than the quantity of the RNA dose by weight. This has been determined (and continues to be determined during production of lots) with a validated quantitative PCR assay.”
The guidelines do not speak of residual DNA limits in terms of their proportion to the amount of RNA, indeed it is unclear why this ratio would be helpful, as surely any risk associated with of DNA would be independent of the amount of RNA.
The EMA do appear to regard this ratio as useful for specification purposes, confirming (116) that the “limit for residual DNA in Comirnaty active substance is 330ng DNA/mg RNA. This equates to <10 ng DNA per adult dose (30 micrograms RNA) of Comirnaty.” This computes to a ratio of 3030:1 of RNA to DNA by weight, consistent with Dr. Marks’ statement that the “ presence of DNA fragments […] is less than three orders of magnitude lower than the quantity of the RNA dose by weight.”
As discussed below ( 6.3 ), the representation of this sort of ratio is highly misleading due to differences in methods use to assay the DNA and RNA.
Question 82: Why does FDA consider the ratio of residual DNA to the amount of RNA relevant in determining the absolute risk of residual DNA in modRNA vaccines? Is this ratio used in the setting of specifications for Drug Substance or Drug Product? What is this specification? Q82
6.2. Guidelines on additional considerations for amount of residual DNA
Dr. Marks stated: “ The agency has taken into account the totality of the mRNA COVID-19 vaccine product, including the lipid nanoparticles, as it reviewed the manufacturers’ specifications for residual DNA fragments present.”
This statement is inconsistent with the documents cited. On the one hand, as described above, the documents cite a residual DNA limit of 10 ng/dose. On the other hand, one of the cited documents ( FDA 2010 (63) ) , as well as others provide for adjustments according to particular circumstances. The 2013 WHO recommendations (50) (p10/110) regarding animal cell cultures as substrates, describe “ recommendations for acceptable levels of residual cellular DNA are product specific .” (emphasis added)
At the heart of the matter is the fact that LNP packaging increases nucleic acid transfection tens to hundreds of times more extensively than for naked nucleic acid. (91-93)
However, the downgrading (see 4.1 ) of the integration risk reflected in the WHO 2021 document (48) is restricted by the experience of DNA vaccines with limited distribution and persistence, noting (p 13/54) that there “ is a wealth of evidence that DNA vaccines to date do not persist or even biodistribute throughout the body of the vaccine recipient when delivered parenterally into muscle,”
Accordingly, these comments cannot be used to dismiss my concerns related to a highly efficient and widespread LNP transfection, nor the principles embodied in the earlier 2007 version (49) of the (48) document: [19] “There would be a need to reassess integration if there was a significant change in the method of delivery, especially any change potentially involving an increase in the capacity of plasmid DNA to enter the nucleus.” (p18/25) (see also 4.4 )
This would certainly be true of a LNP, especially one that contains an SV40 nuclear localization sequence (see 4.2 ). This WHO 2007 document, (49) in the context of discussing non-clinical programs (p20/25), gives an example of electroporation as a “ technique used to improve the uptake of the plasmid.” Electroporation is discussed by Wang et al. (117) and cited by the guideline in another context, also by FDA’s 2007 guidance (64) . [20] Wang et al., showed that electroporation could increase plasmid tissue levels in mice by six- to 34-times and the level of plasmid associated with genomic DNA. In this example, DNA limits might be reduced by a factor of between six and 34.
Question 83: What is FDA’s estimate of the fold-increase of transfection for nucleic acid achieved by the LNPs used in the Pfizer and Moderna modRNA COVID-19 vaccine formulations? Q83
Question 84: Per Question 83, Is this estimate based on FDA’s own studies? If so please describe those studies? If not, was this based on data provided by Pfizer and Moderna? Please provide details. Q84
Dr. Marks’ cited FDA 2010 (63) document provides an example calculation whereby the recommended DNA limit is raised, based on uptake that is less efficient for orally, rather than parenterally administered DNA:
“ You should limit residual DNA for continuous non-tumorigenic cells, such as low-passage Vero cells, to less than 10 ng/dose for parenteral inoculation as recommended by WHO [original Ref. 31 ] . Because orally administered DNA is taken up approximately 10,000-fold less efficiently than parenterally administered DNA, we recommend limiting DNA to less than 100 μg/dose for oral vaccines [original Ref. 32 ] ” (p40/50)
In this case with an assumed fold-decrease in efficiency of 10,000, the DNA limit of 10 ng/dose for a parenteral dose is raised to 10,000 x 10 ng = 100 μg/dose.
Question 85: If FDA permits an upward adjustment in the residual DNA limit in a case where less risk is perceived (i.e. oral dosing), what is FDA’s rationale for not downwardly adjusting the residual DNA limit, in cases where there is more reason to be concerned (i.e. enhanced transfection using LNPs)? Q85
Both the FDA 2007 (64) (p10/13) and WHO 2007 (49) (p23/25) documents addressing DNA plasmid vaccines but involving principles applicable to residual DNA, discuss integration risk in terms of number of DNA copies per μg of tissue, their distribution and persistence. Others have provided related calculations from which to estimate risk. ( (51-53) cited in (50) )
Question 86: What animal or human studies has FDA conducted or solicited from Pfizer or Moderna concerning the biodistribution of residual DNA, quantified in terms of number of copies? Please provide. Q86
Question 87: What algorithms does FDA use to compute integration risk based on the number of copies and sizes of DNA fragments, their distribution and persistence? Please provide details and a record of the calculations performed, Q87
Question 88: How does FDA characterize any possible integration risk for the purposes of determining “safe” exposure levels? For example, does FDA consider exposure to integration-competent DNA capable of producing an (mostly) irreversible effect similar to exposure to ionizing radiation, or rather as an exposure to a toxin that produces a concentration dependent reversible effect? Q88
Question 89: What algorithm does FDA use to adjust the limit of residual DNA per dose, based on FDA’s characterization of risk (per Question 88), the pharmacokinetic properties of residual DNA within LNPs, the interval between multiple doses of COVID-19 vaccine, the interval and dose between the administration of conventional DNA-containing vaccines or non-COVID-19 modRNA vaccines that may be introduced in the future? Q89
6.3. Guidelines on measurement of residual DNA
Dr. Marks stated: “ There are internationally agreed upon recommendations for the quantity of residual DNA present in all biological products, including the mRNA vaccines. [Marks footnote 4 citing (115) , (63) ] The specification for the COVID-19 mRNA vaccines for residual DNA following DNAse treatment results in the presence of DNA fragments at a quantity that is less than three orders of magnitude lower than the quantity of the RNA dose by weight. This has been determined (and continues to be determined during production of lots) with a validated quantitative PCR assay [qPCR] .”
This statement is misleading for several reasons:
The cited WHO and FDA documents do not specify a test method for residual DNA
· Although the FDA 2010 (63) document mentions PCR as a test method in other contexts, it does not specify a method to quantify residual DNA.
· The cited WHO 2007 (115) document states: “With respect to the specification for residual cellular (rc) DNA in the final products, it has been considered that a 10 ng/dose was appropriate for biologicals produced in continuous cell lines. This specification is not associated with an analytical method , and thus additional points should be taken into consideration.” (p20/30) (emphasis added)
This document states that the DNA limit should be defined based on, inter alia, the assay method: (emphasis added) “A risk assessment should be done in order to define the DNA upper limit for a particular vaccine or biological product, based on the following parameters: nature of the cell substrate, inactivation process, the method used to assess DNA content , and the size distribution of DNA fragments.” (p20/30) The document does recognize the value of qPCR as a “highly sensitive and reproducible technique,” (p21/30) but in connection with fragment size analysis, results should be adjusted for amplicon length and amplification efficiency.
Question 90: Please describe the method used to adjust raw estimates of residual DNA for amplicon length and amplification efficiency. Q90
The statement misleadingly implies that qPCR is also used to estimate RNA
Referring to the use of qPCR to determine the excess of RNA over DNA by three orders of magnitude is misleading because it also implies that RNA is similarly measured. This does not appear to be the case, as EMA have confirmed (116) that although qPCR is used to determine residual DNA in Drug Substance, RNA is assayed by UV spectroscopy in the DS and by a fluorescence assay test method in the DP.
Question 91: Please confirm which test methods are used to determine RNA and residual DNA in Drug Substance and Drug Product. Q91
The statement is misleading because qPCR underestimates the amount of residual DNA
It is well known that a qPCR method underestimates the amount of residual DNA. Moderna’s 2018 US Patent 10,077,439 B2 titled “ Removal of DNA fragments in mRNA production process” explains:
“Quantitative PCR is often applied to measure the residual DNA but it only detects the DNA molecules that contain both qPCR primers thus does not measure all other smaller DNA molecules that are partially digested.”
Just as UV and fluorescence methods detect small and large fragments of RNA, they can be used similarly for DNA, especially for fragments substantially smaller than the amplicon length used in PCR. The amount of DNA estimated by these methods can exceed the amount estimated by qPCR by multiples of tens or hundreds. The use of UV absorbance at 250 nm to method to estimate plasmid quantity is referred to in a 2021 WHO guideline. (48)
Question 92: Please provide a justification for why UV or fluorescence methods have not been used to determine the amount of residual DNA, as they appear to be used to estimate RNA. Q92
Question 93: What are the sequences and lengths of amplicons used in the “validated quantitative PCR assay” you refer to that is used to estimate the amount of residual DNA? Q93
Question 94: For both Moderna and Pfizer, what is the smallest length of DNA that can be detected by the particular amplicons used, and under the assay conditions used, for the “validated quantitative PCR assay” used to estimate residual DNA? Q94
Question 95: What studies have FDA performed or solicited from Pfizer or Moderna to characterize the size distribution of residual DNA fragments as a function of amplicon length? Please provide. Q95
Question 96: What is the percentage of total residual DNA detected by qPCR? Q96
Question 97: Please supply the results of residual DNA assay in DS, or DP, for all lots of Pfizer-BionTech or Moderna EUA or BLA COVID-19 vaccines. Please provide the total number of doses supplied, and if known, administered, of each lot, within the USA. Please supply the date of first release for each lot. Q97
Question 98: What measures have been taken to reduce the level of residual DNA contamination? Q98
“three orders of magnitude” is misleading because RNA and DNA are assayed by different methods
As discussed in 6.1 , the EMA describes the residual DNA limit in terms of a RNA-to-DNA ratio of 3030:1, consistent with Marks’ statement “three orders of magnitude.” This is extremely misleading, since DNA and RNA are assayed by different methods, with the qPCR method underestimating the amount of DNA.
Question 99: What studies have FDA performed or solicited from Pfizer or Moderna to characterize the differences between qPCR and UV or fluorescence methods of estimating the amount of residual DNA? Q99
Question 100: Please supply the test protocols for estimating DNA or RNA by qPCR, UV absorption or fluorescence methods, including details of sample preparation to ensure recovery from LNPs and the reduction of confounding of RNA measurements by DNA, or vice-versa. Q100
Question 101: Please confirm that residual DNA is measured at the end of the IVT process, and not in the final drug product. Please justify why it should is not also measured in the final DP formulation, commenting on whether there is free DNA outside of the LNP. Q101
6.4. Guidelines on fragment size of residual DNA
6.4.1. What do the guidelines say about fragment size of residual DNA?
Both documents cited by Dr. Marks regarding “ internationally agreed upon recommendations for the quantity of residual DNA” describe the need to characterize the size distribution of residual DNA fragments, and to limit the fragment size to below 200 base pairs (emphasis added):
FDA 2010 (63)
· “You should measure the amount and size distribution of residual DNA in your final product.” (p40/50)
· “The risks of oncogenicity and infectivity of your cell-substrate DNA can be lessened by decreasing its biological activity. This can be accomplished by decreasing the amount of residual DNA and reducing the size of the DNA (e.g., by DNAse treatment or other methods) to below the size of a functional gene (based on current evidence, approximately 200 base pairs ).” (p40/50)
WHO 2007 (115)
· “Studies performed at CBER suggest that DNA fragments smaller than 200 bp will give substantial safety margins for products that meet the 10 ng per dose limit. This raised some questions concerning the feasibility of detecting small DNA fragments and of quantifying the proportion of the digested DNA of different sizes.” (p18/30)
· “Discussion on the determination of small DNA fragments led to the conclusion that quantitative amplification methods can detect DNA fragments with sufficient sensitivity. In addition, it was concluded that in order to determine the percentage of fragments within a certain size range, both fragments smaller than 200 bp and those larger than this value should be measured and compared , adjusting results for length of the amplicon (and thus amplification efficiency). It was recognized that qPCR is a highly sensitive and reproducible technique.” (p21/30)
· “A risk assessment should be done in order to define the DNA upper limit for a particular vaccine or biological product, based on the following parameters: nature of the cell substrate, inactivation process, the method used to assess DNA content, and the size distribution of DNA fragments.” (p20/30)
6.4.2. Is fragment size of residual DNA being measured?
Despite citing these documents, Dr. Marks’s response is silent on the issue of fragment size distribution.
Question 102: Are residual DNA fragment size or size distribution critical quality attributes for modRNA DS or DP? What methods are used to determine fragment size and distribution? Q102
Question 103: Are residual DNA fragment size or size distribution critical quality attributes included in release specifications? Q103
Question 104: Are residual DNA fragment size or size distribution determined as part of the lot release requirements? Q104
Question 105: According to a FOIA disclosure from Health Canada p24/584 in (118), Pfizer claimed they had never been asked by any regulator to conduct a DNA fragment size distribution analysis. Please confirm. If true, please justify. Q105

Question 106: Did Pfizer provide a DNA fragment size distribution analysis? Please provide. Otherwise please explain why they were not asked to do so. Q106
Question 107: Did Moderna provide a DNA fragment size distribution analysis? Please provide. Otherwise please explain why they were not asked to do so. Q107
6.4.3 Fragments of
As described in section 5 , fragments of residual DNA larger than 200 bp have been found in modRNA vaccines by more than one laboratory. Mean and maximum fragment sizes of 214 bp and 3500 bp respectively ( Figure 6 ) were reported by Speicher et al. (3) using Oxford Nanopore Sequencing. Dr. Phillip Buckhaults reported finding fragments up to 5000 bp. (10) ( Figure 7 )
Question 108: What studies has FDA conducted or solicited from Pfizer or Moderna to describe the fragment size distribution of residual DNA in modRNA vaccines? Please provide methodological details. Q108
Question 109: What percentage of lots of COVID-19 modRNA vaccines failed release testing either by manufacturers or FDA because fragment size criteria were out of specification? Q109
Question 110: If fragment size data were not part of release criteria, but nonetheless measured, what percentage of released lots of COVID-19 modRNA vaccine contained fragments larger than 200 bp? Please stratify by manufacturer, presentation (adult vs. children’s dose etc.), and variant type (original, bivalent, XBB.1.5). Q110
Dr. Marks’ cited WHO 2007 (115) document refers to work at FDA’s CBER that “ DNA fragments smaller than 200 bp will give substantial safety margins for products that meet the 10 ng per dose limit.” (p18/30) This implies that the same safety margin at the same 10 ng dose limit will not exist in lots containing fragments larger than 200 bp.
Question 111: Given the finding that released lots did contain fragments of residual DNA greater than 200 bp and given the above statement in the cited WHO 2007 (115) document, what adjustments to the 10 ng dose limit are required to preserve the same safety margin? Q111
The premise underlying the 200 bp size limit described in the FDA 2010 (63) document, is that lengths below this are considered to be “ below the size of a functional gene.” (p40/50)
In the above cited work (3) and related work, intact sequences of the SV40 enhancer-promotor-ori and the kanamycin resistance gene used as a selection marker in the plasmid process were found, indicating a failure in the DNase digestion step (see 3.4 ).
Question 112: What studies has FDA conducted or solicited from Pfizer or Moderna to determine the prevalence of intact sequence elements from the plasmid vectors in the pool of residual DNA found in COVID-19 modRNA vaccines? Q112
See further questions in section 7.1 and Question 127 and Question 128 .
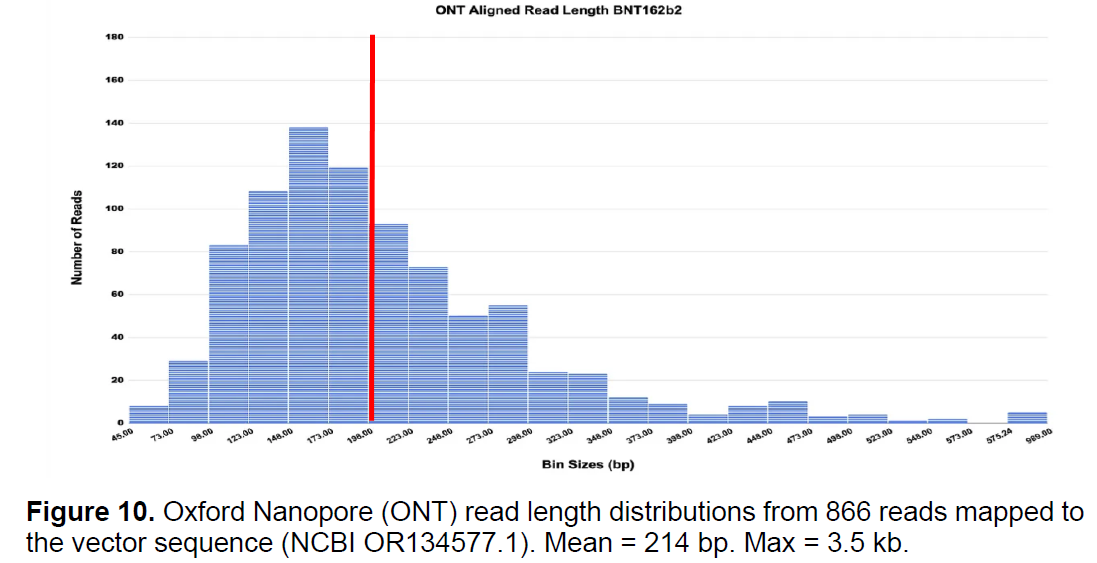
Figure 6 : Fragment size analysis from (3) .
Red line added indicating approximate position of the 200 bp guideline
6.4.4 Concerns
The 200 bp guideline in FDA 2010 (63) is linked to a concern that a functional DNA sequence may integrate into the genome: (excerpted):” The risks […] can be lessened by […] reducing the size of the DNA […] to below the size of a functional gene (based on current evidence, approximately 200 base pairs).” (p40/50)
Indeed, the 10 ng/ dose limit appears to be linked to the 200 bp size limit in the WHO 2007 (115) reference to work at FDA’s CBER that “ DNA fragments smaller than 200 bp will give substantial safety margins for products that meet the 10 ng per dose limit.” (p18/30)
This linkage of fragment size to dose to risk related to insertion of a functional gene might explain the apparent non-concern that qPCR, as discussed above will fail to detect small fragments (see Question 94 ), perhaps those smaller than approximately 100-200bp. Importantly this linkage ignores other risks of DNA that are independent of fragment size (see 4.6 ) and insertional mutagenesis. (49) FDA’s Sheng-Fowler et al., (65) posit two mechanisms for DNA -induced oncogenesis through integration:
· Activation or increased expression of a proto-oncogene such as c-myc, should integration occur next to the proto-oncogene.
· Functional Inactivation of a tumor-suppressor gene, such as the p53 gene (119) or the RB gene, should DNA integration occur within that gene.
Similarly, depending on the site of insertion, integration could interfere with the function of other genes, and in a manner independent of fragment size.
In their 2010 review of FDA guidance on DNA vaccines, authors mostly from the National Cancer Institute (120) noted “ the technology used to detect plasmid persistence does not examine the frequency with which short fragments of plasmid integrate. In this context, sections of DNA as short as 7 bp can affect rates of integration or recombination.”
In his testimony to a committee of the South Carolina Senate, Dr. Phillip Buckhaults (10) stated that “ the probability of a DNA piece of DNA integrating into the human genome is unrelated to its size.”
Figure 7 : DNA fragment size analysis reported by Dr. Phillip Buckhaults (10) . Red line added indicating the approximate position of a peak at about 100 bp
Dr. Buckhaults further testified that “ your genome risk is just a function of how many particles there are,” and noted that the “pieces are very small because during the process they chopped them up to try to make them go away but they actually increased the hazard of genome modification in the process.”
As can be seen from Figure 6 and Figure 7 a substantial fraction of residual DNA is smaller than the 200 bp size limit, representing a substantial potential risk that has not been considered.
The apparent discounting of the possible risks of small fragment residual DNA serves only to heighten concerns expressed in earlier questions, particularly Question 71 and Question 69 .
The relief of earlier concerns about plasmid DNA expressed in the WHO 2021 guideline (48) may have been based, partly on the notion that the “local response to plasmid DNA inoculation is that cells take up the plasmid and then express the immunogen(s) encoded in the DNA vaccine and/or the nucleic acid is degraded by normal molecular mechanisms.” (p14/54) This degradation may have been considered to be more rapid for naked DNA shorter than 200 bp. These mitigating factors cannot be assumed to apply to residual DNA that could be encapsulated and protected by LNPs,(see p21/66 in (71) ) or that may form RNA hybrids that are resistant to endonucleases, or the possibility of DNA-lipid adducts (Section 8 ).
The 2007 FDA guidance on plasmid vaccines articulates a principle that is applicable to modRNA vaccines:
“If the DNA sequence of the insert gene and/or backbone vector of a DNA vaccine are changed, we recommend that you consult with CBER to discuss whether the nature and/or magnitude of the change(s) warrant the conduct of additional preclinical studies and/or the submission of a new IND. You should provide to CBER a description of the changes in manufacturing process and the results from preclinical safety evaluations of the new (modified) DNA vaccine.” [p5/13]
Question 113: Please summarize and tabulate all changes to the sequences of the DNA plasmid vector and the modRNA DS used in the preclinical tests, clinical studies, and post-authorization to the current versions of COVID-19 vaccines. Please indicate the reason for each change and what analytical, non-clinical or clinical comparability studies were performed to qualify these changes. Q113
Question 114: Per Question 113, if no preclinical or clinical studies were performed for any given change, please provide a rationale. Q114
7. Concerns regarding intact plasmid sequence elements
There are several concerns related to these sequences, particularly as they have been found as intact sequences.
7.1. Antibiotic resistance gene
Valera et al. noted that antibiotic resistance genes used in the infection or transfection protocols may, if introduced into mammalian cells exert a biological effect: “neo gene expression may induce changes in the cells, which should be considered when neo-selected cells are used to deliver specific genes in different therapy approaches and in embryo manipulation.” (121)
The WHO 2007 guideline (49) on plasmid DNA vaccines states:
“If other gene constructs are included in the plasmid, such as antibiotic resistance genes for manufacturing reasons, then the possibility of expression of such gene sequences in mammalian cells or in microorganisms which are potentially pathogenic, and the possible clinical consequences of such expression, should be considered.” (p20/25).
Question 115: What risk-assessment or studies have FDA conducted or solicited from Pfizer or Moderna related to the transfection of an antibiotic resistance gene within residual DNA into a vaccinee? Q115
Question 116: What risk-assessment or other studies have FDA conducted or solicited from Pfizer or Moderna related to the transfection of antibiotic resistance genes into commensal or infection pools of bacteria in a vaccinee? Q116
Question 117: What risk-assessment or studies have FDA conducted or solicited from Pfizer or Moderna related to the transfection of antibiotic resistance genes into commensal or infection pools of bacteria in a vaccinee? Q117
Question 118: What risk-assessment or studies have FDA conducted or solicited from Pfizer or Moderna related to the transfection of antibiotic resistance genes into environmental (e.g. soil, wastewater) bacteria? Q118
In the study performed by high school students under FDA supervision (11) replication competent DNA from “in-house mRNA” containing the antibiotic-resistant gene were detected. It is possible that this was in fact manufacturer supplied vaccine.
Question 119: Please provide details of the “in-house mRNA” used by Wang et al., (11) particularly its source and similarity to EUA of BLA material. Please provide all raw data for this study, and describe the involvement of FDA staff and their relationship to the student. Please provide the protocols or other documentation likely needed for submission to the R&D committees that would have been needed to approve the conduct of the study. Q119
7.2. SV40 promoter-enhancer and other sequences
7.2.1 Concerns about
Dr. Marks stated: “No SV40 proteins are encoded for or are present in the vaccines.” The authors of the studies (2, 3) describing SV40 sequences in the modRNA vaccines do not assert that SV40 proteins are present in the vaccine. None of my questions concern the expression of SV40 proteins, as the sequence concerned is a regulatory element that is not acting via protein expression.
There are debated associations between SV40 virus and cancer (122) particularly regarding contaminated polio vaccines in the 1960s and the role of the SV40 Large T antigen oncoprotein. (123) Another chapter (Tumor viruses) in the textbook (57) cited by Dr. Marks (see 4.2 ) discusses genomic integration with DNA viruses such as the SV40 virus. FDA’s own staff acknowledge that SV40 virus contains “potent viral oncogenes” (97)
7.2.2. Possible consequences of the SV40 enhancer-promotor-ori sequences
The above cited 2010 review of FDA guidance on DNA vaccines from the National Cancer Institute (120) observed that “in evaluating the potential harm of plasmid integration, it should be noted that the risk of introducing plasmids with strong regulatory regions into the host genome far exceeds that associated with random point mutations.” (emphasis added) The SV40 enhancer=promoter-ori sequence would certainly qualify as a “strong regulatory region.”
The nuclear localization properties of this sequence have been discussed above (see 4.2 ). Additionally this sequence binds the p53 protein (119) which has both tumor suppressor (119) and antiviral (124) properties. Sequestering p53 may therefore have consequences in terms of oncogenesis and viral pathogenicity. The SV40 promoter may also enhance expression of episomal DNA transgenes, without need ing insertional mutagenesis" (101) (see 4.5 ). See Question 127 and Question 128 .
7.2.3. Was the SV40 and other sequence and other sequences identified to FDA?
The FDA 2010 guidance (63) cited by Dr. Marks addresses the necessity of providing information about plasmids used in vaccine production: “Whatever starting materials are used for the generation of the cell substrate (e.g., parent cells or plasmids used for genetically engineered cells), any available information about those starting materials and their characterization (e.g., sequence of the plasmid ) should be provided .” (p9/50) (emphasis added)
A 2021 WHO document (71) on mRNA vaccines provides more detail on this guideline, at least for the DNA template: “The annotated sequence of the DNA template should be provided. The sequence and position or length of all elements contained within the mRNA, including start and stop codons, flanking UTRs, regulatory elements (for example, promoter for the RNA polymerase) and 5′ cap and 3′ poly(A) tail, should be provided, as well as the ORF for the target antigen. If any additional proteins are encoded (such as those for a self-amplifying construct or a cytokine) their sequence should be provided (see points d and e below). The presence and function of any additional sequences included in the construct should be described.” (p24/66)
However, in the absence of further details in the 2010 FDA document, (63) the FDA 2007 guidance regarding plasmid DNA vaccines. (64) furnishes broadly applicable principles regarding the disclosure of the nature of the plasmid (p4/13) that manufacturers “ should provide detailed descriptions of the plasmid construction, including the source and diagrams of all plasmids used […] along with an annotated sequence identifying all open reading frames including any unexpected open reading frames and/or other sequence elements .” (emphasis added) WHO guidelines from 2007 (49) (p8/25) and 2021 (48) (p18/54) contain similar language.
In a response to the Epoch Times, Health Canada stated: “Health Canada expects sponsors to identify any biologically functional DNA sequences within a plasmid (such as an SV40 enhancer) at the time of submission. Although the full DNA sequence of the Pfizer plasmid was provided at the time of initial filing, the sponsor did not specifically identify SV40 sequence .”
The need to provide the details of plasmid construction as part of the manufacturing and quality review is self-evident. The plasmid map appeared in an EMA assessment report, (125) shown below and compares Pfizer’s disclosed plasmid map with that generated by Illumina sequencing by McKernan et al. (2, 3) In this graphic, the orientation of the EMA-disclosure sequence has been flipped to facilitate comparison between the two maps.
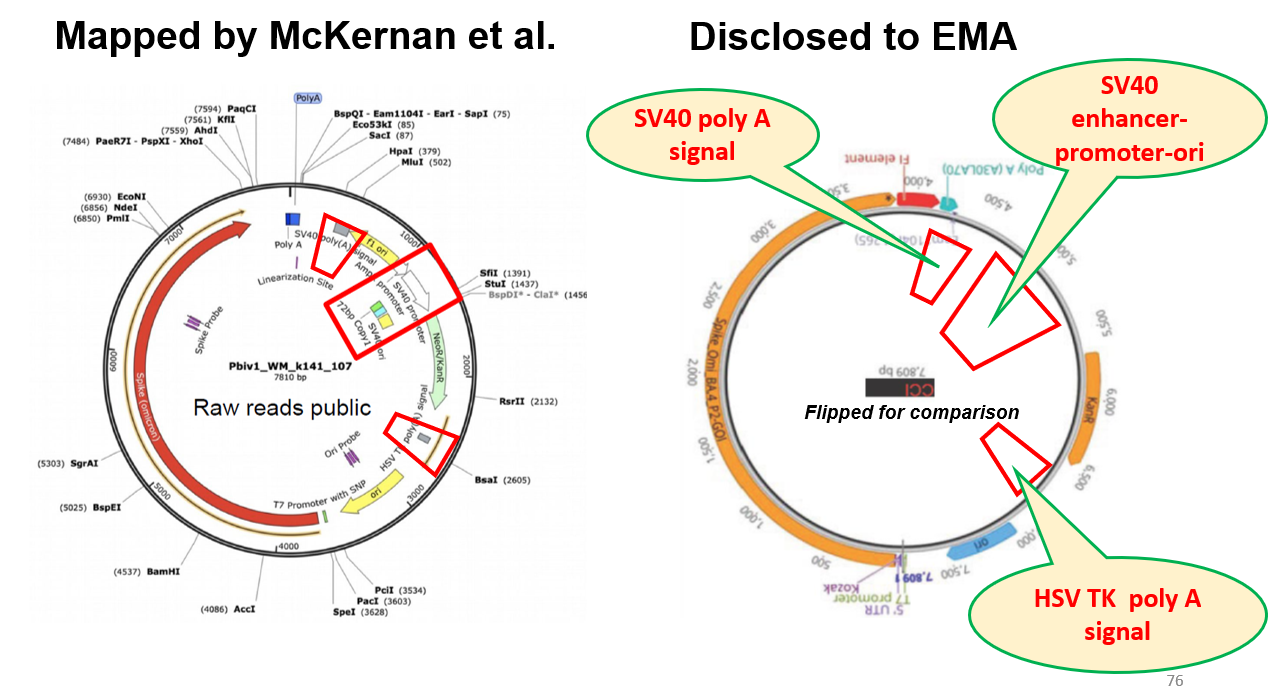
Figure 8 : Comparison of plasmid maps obtained by McKernan et al., and as disclosed to EMA
The red boxes show that three sequences were not disclosed to the EMA: 1) SV40-promoter-enhancer-ori, 2) SV40 poly(A) signal; 3) HSV-TK poly(A) signal. These are all regulatory sequences. There are two other features of note:
· The AmpR promoter which is a regulatory sequence required for the expression of the antibiotic resistance gene needed for production of the plasmid, is missing in the Pfizer version.
· Software, such as Snapgene, generates these plasmid maps within seconds of loading the sequence, showing all the sequence elements, as was the case in the McKernan version. It appears that using this software for the three undisclosed sequences to be absent from the Pfizer version would have likely have required editing of the generated map by the selection of a particular element and using the “delete” function to delete it.
In a November 27 2023 response to a posed question (116) , EMA confirmed that: “an SV40 sequence is present in the DNA plasmid starting material of Comirnaty and its adapted vaccines, including Comirnaty Omicron XBB.1.5.”
EMA further noted: “The full DNA sequence of the plasmid starting material was provided in the initial marketing authorisation application for Comirnaty. The MAH [Marketing Authorization Holder] did not specifically highlight the SV40 sequence, as it was considered to be a non-functional part of the plasmid. They have since clarified this information in response to questions raised by EMA.”
It appears from this response that Pfizer’s non-disclosure of these sequences was a purposeful rather than an inadvertent act, having at some point conducted what amounts to a risk assessment regarding this sequence that had not been disclosed to EMA because it “ considered [the SV40 enhancer-promoter sequence] to be a non-functional part of the plasmid .” (116) Even if it could be argued that FDA guidances for plasmid-based DNA vaccine cannot be applied to modRNA vaccines made from plasmid templates, submission of a plasmid map from which certain sequences have been removed without remark appears to constitute a false or misleading disclosure.
A FOIA disclosure from Health Canada (118) confirmed this:
“Pfizer has communicated to us recently, that they apparently chose not to mention this information to EMA, FDA or HC at the time of t heir initial or subsequent submissions. However, as of April of this year this information was independently made public, which has resulted to questions coming to agencies.” (p162/584)
Question 120: Please confirm that the plasmid template used to produce all Pfizer-BioNTech COVID-19 vaccines made under EUA or BLA, to date (including the XBB.1.15 vaccine) contain sequences for 1) SV40-promoter-enhancer-ori, 2) SV40 poly(A) signal; 3) HSV poly(A) signal. Q120
Question 121: Please describe FDA’s expectation, by statute, regulation, or practice for sponsors to disclosure all sequence elements contained in the plasmid template used for the production of modRNA or mRNA vaccines. Q121
Question 122: Please describe whether Pfizer disclosed the full plasmid sequence of its plasmid to FDA and whether this disclosure included specific details of sequence elements, including the three sequences listed above apparently not disclosed to EMA or Health Canada. Please describe whether these disclosures included an annotated plasmid map. Please answer this question for all variant (Wuhan, bivalent, XBB1.5) vaccine versions, whether under EUA or BLA. Please provide the dates of disclosure of the full sequence and the details of any sequence elements not disclosed along with the full sequence. Q122
Question 123: If these three sequence elements were not detailed at the same time as the full sequence, please provide Pfizer’s justification for not doing so. Q123
Question 124: Please provide the date when FDA asked Pfizer whether or these sequences were present in their plasmid. Q124
In the above referenced inquiry, (116) EMA were also asked to provide any risk analysis conducted by EMA or Pfizer regarding these SV40 sequences. EMA responded: “ A detailed risk assessment and further supporting data has been requested from the company and will be assessed by EMA/Rapporteurs.” From this it appears that Pfizer had not heretofore submitted the risk-assessment related to these sequences they had apparently conducted when, according to the EMA, they had not pointed out these sequences because they had considered them to be non-functional.
What makes the absence of any risk assessment regarding these sequences all the more egregious is the fact that this must have been omitted from a risk assessment submitted to EMA relating to process-related impurities, as memorialized in the EMA Assessment of February 19 2021 (30) (p19/140): “A safety risk assessment for potential process-related impurities included in the active substance process relative to patient safety was performed. The sources of the impurities are sufficiently addressed. The safety risk assessment strategy involves comparison of the theoretical worst-case concentration of impurities, assuming no removal, to calculated safety concern thresholds.”
Question 125: Please describe whether and when Pfizer disclosed to FDA the function of these three sequences. Q125
Question 126: Please state when FDA asked Pfizer to describe the function of these sequences. Please describe the function of these three sequences. Q126
Question 127: Please state whether Pfizer or FDA provided or performed a risk assessment related to the presence of these sequences, as intact sequences in residual DNA in Drug Product? If one has been submitted or prepared, please provide a copy. Q127
Question 128: Per Question 127, does this assessment consider the actions of the SV40 enhancer-promoter-ori described in section 7.2.2? If not, please discuss these topics. Q128
Question 129: Please state when FDA asked Pfizer to provide a risk assessment related to these sequences. Q129
The cited FDA 2010 guidance (63) states: “The risks of oncogenicity and infectivity of your cell-substrate DNA can be lessened by decreasing its biological activity. This can be accomplished by decreasing the amount of residual DNA …” (p40/50) (emphasis added)
Question 130: If, according to the FDA 2010 guidance (63)the risks of DNA can be lessened by reducing the amount of residual DNA, please provide a justification for increasing the load of DNA by the inclusion of SV40 sequences that are, according to EMA, (116)“non-functional.” Q130
Question 131: If, per the above questions, Pfizer failed to make the appropriate disclosures regarding the presence, function or assessment of risk of these sequences in a timely fashion, what regulatory actions were and will be taken against Pfizer? What was Pfizer’s justification for failing to make these disclosures? Q131
The cited FDA 2010 guidance (63) refers to the CFR to define “extraneous material:” (emphasis added)
“The regulations, in 21 CFR 610.13, [21] state in part that “Products shall be free of extraneous material except that which is unavoidable in the manufacturing process described in the approved biologics license application.” In 21 CFR 600.3(r), [22] purity is defined as the “relative freedom from extraneous matter in the finished product, whether or not harmful to the recipient or deleterious to the product .” (p6/50)
Question 132: If the SV40 sequences are indeed non-functional,” and their inclusion not unavoidable, it would appear that intact or fragmented SV40 or HSV sequences found in residual DNA constitute “extraneous material.” What investigative or enforcement actions has FDA taken to correct this apparent violation of the regulations that ““Products shall be free of extraneous material.” Q132
Pfizer did provide to Health Canada (126) an risk assessment of the SV40 sequences that stated:
“Importantly, the SV 40 sequence elements are not oncogenes and do not cause cancer [7]. As described above (See Section 3.1, #4) residual DNA is expected to degrade rapidly and has a very low likelihood of reaching the nucleus. In the unlikely theoretical event that the SV40 promoter/enhancer and the [REDACTED] elements would reach the nucleus intact and transient expression of the resistance gene occurs, this would have no biological effect to the vaccinee. Therefore, these sequence elements do not pose any safety risk to the vaccinee.”
Question 133: Did FDA receive a document similar to that provided to Health Canada? (126) When? Please supply unredacted text. Q133
Question 134: What evidence did Pfizer present to justify the statement: “residual DNA is expected to degrade rapidly “ Did FDA ask Pfizer to provide this evidence? Please provide. Q134
Question 135: What evidence did Pfizer present to justify the statement: “residual DNA […] has a very low likelihood of reaching the nucleus. “ Did FDA ask Pfizer to provide this evidence? Please provide. Q135
Question 136: Did Pfizer quantify, with justification, just how likely or unlikely the sequences described could reach the nucleus? Did FDA ask Pfizer to provide this evidence? Please provide. Q136
Question 137: Given the absence of a nuclear member in mitosis ( 4.2), and the ability of the SV40 sequence to act as a nuclear localization signal, (69, 127) did FDA challenge Pfizer on the assertion that “residual DNA […] has a very low likelihood of reaching the nucleus.“Please provide. Q137
Question 138: Did Pfizer quantify, the likelihood of expression of the resistance gene, as well as the duration of its “transience.” Did Pfizer describe what biological this gene would have if expressed and explain why this would not pose a safety risk? Did FDA seek answers to these questions? Q138
Question 139: Has FDA asked Pfizer to remove the SV40 sequences from their plasmids? What is the schedule for this? Ny what regulatory pathway will these non-SV40 versions of Pfizer’s pro-vaccines be approved? Will RCT’s be required? Q139
7.3. Unintended sequences in DNA plasmid vector
The need to investigate the presence of “unexpected open reading frames” or “ unintended sequences of biological significance” in DNA plasmids is described in the 2007 FDA guidance (64) (p4/13) and the 2021 WHO guideline (48) (p18/54) respectfully.
Given the opportunity for large residual DNA fragments (see 6.4.3 ) precaution should be equally prudent in the case of residual DNA in modRNA vaccines.
Question 140: .What investigations were performed by Pfizer, Moderna, FDA, or other government to determine the presence of “unexpected open reading frames” or “unintended sequences of biological significance” in both strands of the plasmid vector used to produce the modRNA COVID-19 vaccines? Q140
Question 141: Please provide the study reports of any investigations performed per Question 140, along with risk assessments related to the findings. Q141
8. RNA or DNA Lipid Adducts , “Process 3”
8.1. Formation of RNA or DNA Lipid Adducts
In 2021 Moderna scientists (128) described the formation of lipid-mRNA adducts in LNPs. In their 2022 Science Day presentation, (129) Moderna revealed that these adducts result from aldehyde formation related to impurities in LNP raw materials. The formation of adducts may stall ribosomal translation, inactivating the entire mRNA molecule. Adducts could be avoided by control of the manufacturing process and by using “aldehyde sinks” such as Tris as buffers instead of the more common PBS (phosphate buffered-saline).
|
|
|
|
Slide 76/136 |
Slide 75/136 |
|
Detail:
|
|
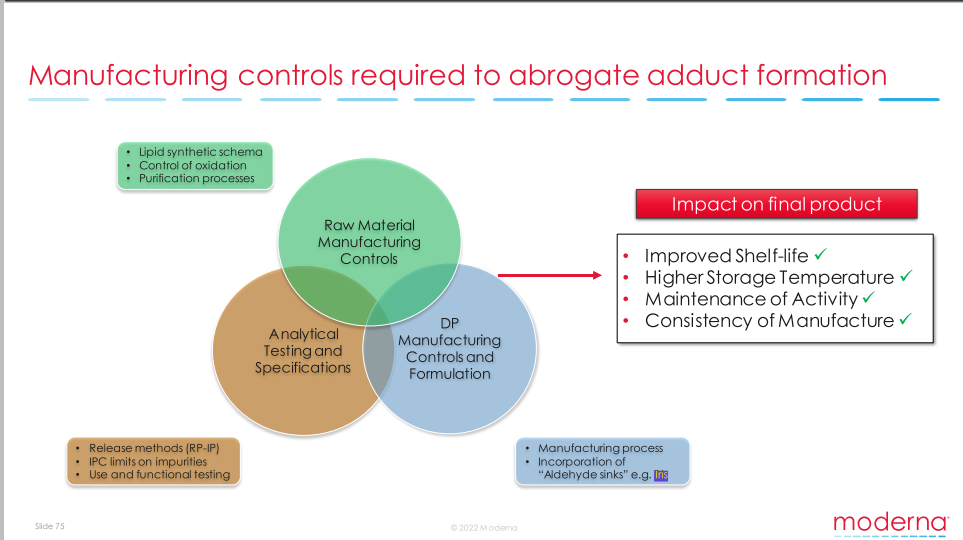
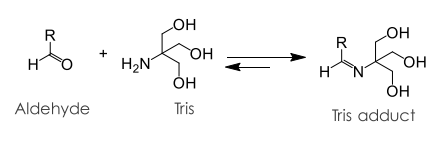
Figure 9 : Slides from Moderna’s 2022 Science Day presentations (130)
This information appears absent from Moderna’s Science Day presentation in 2020. (130) Given that dosing in Moderna’s distribution study was initiated on July 10 2017 (p11/280) and the report approved on December 13 2017 (p25/280) (131) it seems unlikely that this study predated Moderna’s discoveries regarding lipid-mRNA adducts.
During storage, tertiary amine–based LNP lipids can oxidize to form reactive aldehydes that covalently adduct modRNA, blocking its translation after injection. (128) These findings raise concerns that lipid–RNA adducts may contribute to LNP persistence, unmeasured toxicity, and poorly understood long-term biodistribution and toxicity. (132)
Adducted modRNA taken up by cells may be sensed as abnormal or viral-like, triggering interferon signaling and contributing to systemic immune dysregulation.(Cordes, J.; Zhao, S.; Engel, C. M.; Stingele, J. Cellular responses to RNA damage. Cell 2025, 188 (4), 885-900. DOI: https://doi.org/10.1016/j.cell.2025.01.005 )
In addition to their effects on translation, a variety of disorders and diseases may be related to aldehydes. Various DNA adducts may be mutagenic: “Mutagenesis could result from the processing of either DNA adducts or a combination of DNA, RNA, and protein adducts.” (133) Some possible mechanisms have been discussed elsewhere. (134) DNA adducts mentioned in FDA’s 1997 guidance on genotoxicity testing of drugs. (p6/12 in (135) )
In a EMA recommendation (p163/169 in (136) ) Moderna was asked to “provide evidence to confirm that the impurities and/or degradation products resulting from PEG2000-DMG, cholesterol and DSPC have been sufficiently investigated and do not result in the formation of lipid-RNA species by 31-01-2021.”
Question 142: When did FDA become aware that lipids may form adducts with nucleic acids? Q142
Question 143: What is the nature of the lipid-RNA species and why might they be a concern? Q143
Question 144: Did FDA have a similar concern for lipid-RNA species as did EMA? Were these concerns based on formation of aldehyde-related adducts, or other mechanisms? Q144
Question 145: How was this concern lipid-RNA species resolved? Q145
Question 146: Given that the work on the lipid-RNA species was to be provided by January 1 2021, when exactly did this occur? Q146
Question 147: If lipid-RNA species prior to resolution of this issue, how many doses of mRNA-1273 had been administered either in clinical trials or post approval/ authorization? Q147
Question 148: Are there specific guidelines and limits on these adducts? How are they controlled? Did Pfizer and Moderna comply with these guidelines? Q148
8.2. Process 3: Pfizer buffer change
In October 2021, Pfizer changed the buffer in their pro-vaccine product from PBS to Tris, ostensibly to make the cold storage requirements less onerous. FDA authorized this switch based on bench top analytical rather than animal or human trial comparability. Although Moderna had described (see above) the role of buffer on lipid adduct formation, this was not widely known or described to VRBPAC.
More recently (December 2023), a BioNTech patent application (137) describing the advantages of using Tris buffer compared with PBS. The main advantage, different from those given by Moderna but with the same end result, is that the Tris prevents the mRNA strand from folding in a certain way that would otherwise impair its translation. The patent application also describes other modifications to the composition of the LNP buffer solution that along with the Tris, alter the stability of LNPs.
It is difficult to understand why, given the formation of the folded mRNA structures, this sort of process (“Process 3”) change would be suspected of affecting safety and effectiveness and therefore trigger biological comparability studies. (138) Moderna have discussed how buffer may affect a number of LNP, properties, such as size, pharmacokinetics, and expression kinetics. (139) Others have described the effects of different buffers in in vitro and in vivo models on the mRNA transfection efficiency of LNP. (140)
Even more concerning is that BioNTech’s patent application was filed on November 15, 2021, just two weeks after FDA authorized (141) the formulation change on October 29 2021 based on the premise that the new buffer would improve stability and storage cold requirements with no mention that the molecular form of the mRNA would be changed. Yet more concerning is that BioNtech may have known about these problematic molecular forms and the stability of LNPs in different buffer solutions a year earlier, around the time of the original EUA. Table 2 of the patent application (137) provides the dates of some of these experiments – from December 18 2020 to January 22 2021.
Figure 10 : Table 2 from Pfizer’s patent application (137)
Question 149: Given what was known at the time about lipid adducts and their possible biological consequences, what studies analytical, preclinical or clinical studies did FDA require from Pfizer when they changed their buffer? What were the results or requested or voluntarily provided studies? Q149
Question 150: Why were the possible biological consequences of a buffer change fully disclosed to VRBPAC who were being asked to make recommendations based on the totality of scientific evidence available and a consideration of known and potential risks? Q150
9. Novel heterotrimers in bivalent pro-vaccines: Process 2 for Moderna, Process 4 for Pfizer
As we have discussed previously, (142, 143) on August 31 st 2022 FDA issued EUAs for “bivalent” Covid-19 versions of the Pfizer (144) and Moderna (145) modRNA pro-vaccines. Since the spike proteins of the BA.4 and BA.5 variants are identical, (146) these vaccines are said to be “bivalent” because they contain modRNA encoding for the spike proteins of the Wuhan and BA4/5 variants.
The term bivalent is a misnomer. Moderna revealed at the Sept 1st ACIP meeting (142, 147) that their bivalent vaccine elicits the formation of novel spike protein heterotrimers to produce a superior immunological response. This occurs because the modRNA for each variant is loaded into the same LNPs. Transfected cells then produce spike protein monomers of each type, which then aggregate into trimers. Four trimers are possible: homotrimers for each of the Wuhan or BA4/5 variants, plus heterotrimers containing either two Wuhan monomers plus one BA4/5 monomer, or one Wuhan monomer plus two BA4/5 monomers.
Thus, the two heterotrimers are entirely novel. Since they have novel chemistry and pharmacology (according to Moderna), they may also have a novel yet untested toxicology. Based on EMA documents, it is likely that the same occurs for the Pfizer bivalent vaccine.
Indeed, Wagenhauser et al. reported (148) that the “rate of adverse reactions for the second booster dose was significantly higher among participants receiving the bivalent 84.6% (95% CI 70.3%-92.8%; 33/39) compared to the monovalent 51.4% (95% CI 35.9-66.6%; 19/37) vaccine (p=0.0028).”
Figure 11 : Graphical results from Wagenhauser et al. (148)
In March 2022, contemplating that updated versions of the original COVID-19 pro-vaccines would be needed, FDA modified its guidelines for the issuance of EUA. (149) The update stated: “FDA expects that much of the manufacturing process and controls, as well as the facilities for vaccine production, for the modified COVID-19 vaccine will be identical to that of the prototype COVID-19 vaccine.”
Producing these bivalent products requires significant changes in manufacturing that go well beyond their expectations of manufacturing comparability with the original versions. Thus, for Pfizer, the production of their bivalent product should constitute “Process 4,” and for Moderna, “Process 2.”
As we reported, (150) at a meeting between the React19 vaccine injury group and FDA on December 14, 2022, FDA acknowledged that heterotrimer formation is possible with the bivalent vaccines, but claimed that just because heterotrimers may display different antigen properties from homotrimers, their toxicological properties would likely be the same. Despite undertaking to provide further information, this was not forthcoming.
Question 151: By way of tabulation, please compare and contrast, the processes used to produce the original monovalent version of the COVID-19 modRNA provaccines, and the bivalent. Please provide separate comparisons or Moderna and Pfizer. Q151
Question 152: Please provide the questions asked by FDA and the justifications provided by Moderna and Pfizer to support the claim of manufacturing comparability. Q152
Question 153: Were Moderna and Pfizer asked to provide an assessment of toxicological equivalency of the heterotrimer spike proteins to their homotrimer counterparts? Please provide. Q153
Question 154: Were Moderna and Pfizer asked to conduct in vitro, animal or clinical comparability testing, particularly to demonstrate toxicological equivalency of the heterotrimer spike proteins to their homotrimer counterparts? Please provide. Q154
10. Safety studies with modRNA “and lipid nanoparticle together that constitute the vaccine” ?
Responding to concerns about genomic integration of residual DNA into the genome, mutagenesis, and cancer risk. Dr. Marks included two statements regarding the results of genotoxicity studies conducted in animals.
The first statement: “Additionally, animal studies with the mRNA delivery technology done over the past decade show no evidence of genotoxicity.”
This statement provides no details of what studies were conducted or assurance that any of the prototypes tested “over the past decade” were comparable in their formulation, composition of LNPs, plasmid sequence (if used), type of template used, method of manufacture, type of nucleoside modification, codon optimization, sequence of non-coding regions and levels of impurities.
Question 155: Which peer reviewed paper(s) or regulatory document(s), including submissions from Pfizer or Moderna describe the details of “animal studies with the mRNA delivery technology done over the past decade” that “show no evidence of genotoxicity.” Please provide. Q155
The second statement: “Additionally, studies have been conducted in animals using the modified mRNA and lipid nanoparticle together that constitute the vaccine , including the minute quantities of residual DNA fragments left over after DNAse treatment during manufacturing, and demonstrate no evidence for genotoxicity from the vaccine .” (emphasis added) Footnote 3 to this statement contains two URLs, pointing to the “Summary Basis for Regulatory Action” documents for COMIRNATY (47) and SPIKEVAX. (151)
The assertion that these genotoxicity studies were conducted “using the modified mRNA and lipid nanoparticle together that constitute the vaccine” should be read with a related assertion in the FDA same document cited (151) concerning a biodistribution study performed on non-candidate modRNA vaccine of redacted identity.
FDA represents (p14/30) that because the test article was “manufactured using the same procedure as SPIKEVAX” it contained LNPs whose properties matched those of SPIKEVAX, including its “ biodistribution and retention” properties . ( Figure 12 ).

Figure 12 : SPIKEVAX biodistribution: Screenshot (p14/30) from FDA document (151) (highlight added)
Both assertions “using the modified mRNA and lipid nanoparticle together that constitute the vaccine” and “manufactured using the same procedure” provide the foundation for the substitution of directly relevant studies by supportive studies.
A close examination of the formulations used in these studies reveals that t hese assertions are a t best misleading, and in some cases false. It should go without saying that manufacturing procedure and formulation play critical roles in determining the behavior, distribution, and transfection of LNPs, affecting vaccine pharmacology and toxicology. Thus, over-extending extrapolations from supportive studies affects the interpretation and weighing of evidence that has supported the issuance of EUAs and BLAs.
10.1. Which formulations were used in Moderna’s nonclinical safety studies?
The cited document “Summary Basis for Regulatory Action” for SPIKEVAX, (151) contains the passage (p14/30) shown in Figure 13 .

Figure 13 : Screenshot from “Summary Basis for Regulatory Action” for SPIKEVAX, (151)
The number and description of the studies enumerated in this FDA document (151) ( Figure 13 ) do not match with those given in other documents (136, 152) as will be discussed below ( 12.3.3 ).
To determine which formulations were used in the various studies alluded to in the FDA document cited (151) , Table 1 (p43/169) from EMA’s 2021 assessment report (136) is reproduced here ( Table 1 ). The three leftmost columns have been added to facilitate this discussion. Additional abbreviations and footnotes have been added, apparent typos or errors corrected as indicated, with clearer layout. Not included in this excerpt are seven pharmacology studies, of which six were identified as involving mRNA-1273, Moderna’s candidate COVID-19 vaccine. A similar table is given in Moderna’s nonclinical overview (152) (p17/31) from which details related to formulation have been added here as indicated.
Table 1 : Table 1 from 2021 EMA assessment of Moderna vaccine (136) with additions from Moderna (152) and modifications indicated in text
|
ID |
Vaccine |
LNP/ lipid |
Description |
Study # |
Test system |
|
|
Repeat-dose Toxicology Studies |
||||||
|
T1 |
Zika – d mRNA 1706 |
LIP4 |
Zika: A 1-month (3 doses) intramuscular injection toxicity study of mRNA-1706 in Sprague-Dawley rats with a 2-week recovery period d, e m |
5002045 |
SD rat, M and F |
|
|
T2 |
Zika – d mRNA 1706 |
LIP4 |
A 1-month (3 doses) intramuscular injection toxicity study of mRNA-1706 in Sprague-Dawley rats followed by a 2-week recovery period d, e |
5002231 |
SD rat, M and F |
|
|
T3 |
hMPV / PIV3 mRNA 1653 |
LIP4 + PG%% |
A 1-month (3 doses) study of mRNA-1653 by intramuscular injection in Sprague-Dawley Rat with a 2-week recovery period f,e |
5002033 |
SD rat, M and F |
|
|
T4 |
Zika – g (diff from d) mRNA 1893 |
LIP4 + PG%% |
A 1-month (3 doses) intramuscular injection toxicity study of mRNA-1893 in Sprague-Dawley rats followed by a 2-week recovery period g, e |
5002400 |
SD rat, M and F |
|
|
T5 |
CMV – c mRNA1647 |
LIP4 + PG %% |
A 6-week (4 doses) intramuscular injection toxicity study of mRNA-1647 in Sprague-Dawley rats followed by a 2-week recovery period c, e, n |
5002034 |
SD rat, M and F |
|
|
T6 |
CMV (diff from c) $$ mRNA 1443 |
LIP4 + PG %% $$ |
A 6-week (4 doses) intramuscular injection toxicity study of mRNA-1443 in Sprague-Dawley rats followed by a 2-week recovery period i, e $$ |
5002158 |
SD rat, M and F |
|
|
Other Toxicity Study |
||||||
|
T7 |
SARS-CoV-2 mRNA 1273 |
LIP4 |
A non-GLP repeat-dose immunogenicity and toxicity study of mRNA-1273 by intramuscular injection in Sprague Dawley rats a |
2308-123 |
SD rat, M and F |
|
|
Biodistribution Study |
||||||
|
B1 |
CMV – c mRNA1647 |
LIP4 + PG %% |
A single dose intramuscular injection tissue distribution study of mRNA-1647 in male Sprague-Dawley rats c |
5002121 Amendment 1 |
SD rat, M only |
|
|
Genotoxicity Studies |
||||||
|
G1 |
None |
SM-102 only |
SM-102 bacterial reverse mutation test in Salmonella typhimurium and Escherichia coli e |
9601567 |
S. typhimurium and E. coli strains, in vitro |
|
|
G2 |
None |
SM-102 only** |
SM-102 in vitro mammalian cell micronucleus test in human peripheral blood lymphocytes e |
9601568 |
Human peripheral blood lymphocytes |
|
|
G3 |
Zika -d mRNA 1706 |
LIP4 |
Zika mRNA: mammalian erythrocyte micronucleus test in rat d, e m |
9800399 |
SD rat, M and F |
|
|
G4 |
Luciferase model mRNA |
LIP4 ## |
NPI luciferase mRNA in SM-102-containing lipid nanoparticles: in vivo mammalian bone marrow erythrocyte micronucleus assay in the rat ## |
AF87FU.125012 NGLPICH.BTL |
SD rat, M and F |
|
|
G5 |
None |
PEG2000-DMG (b)(4) |
Study added from MODNCO: bacterial reverse mutation test in Salmonella typhimurium and Escherichia coli |
9601035 |
S. typhimurium and E. coli strains, in vitro |
|
|
G6 |
None |
PEG2000-DMG (b)(4) |
Study added from MODNCO: in vitro mammalian cell micronucleus test in human peripheral blood lymphocytes |
9601036 |
Human peripheral blood lymphocytes |
|
|
Reproductive and Developmental Toxicity |
||||||
|
R1 |
mRNA 1273 j |
LIP4 |
Combined developmental and perinatal/postnatal developmental and reproductive toxicity study. (Study Days 1 and 15 [28 and 14 days prior to mating, respectively] and Gestation Days 1 and 13) |
20248897 |
SD rat |
|
Abbreviations : CMV = cytomegalovirus; eCTD = electronic common technical document; ERD = enhanced respiratory disease; F = female; GLP = Good Laboratory Practice; M = male; mRNA = messenger RNA; SM-102 = heptadecan-9-yl 8-((2-hydroxyethyl)(6-oxo-6-(undecyloxy)hexyl)amino)octanoate; Tris- HCl = tris(hydroxymethyl)aminomethane-hydrochloride; VRC = Vaccine Research Centre.
Notes [layout enhanced from original, information from Moderna’s nonclinical overview indicated by MODNCO: ]
a mRNA-1273 contains a single mRNA sequence that encodes for the full-length SARS-CoV-2 S-2P combined in a mixture of 4 lipids (SM-102, PEG2000-DMG, cholesterol, and DSPC) and formulated in 20 mM Tris, 87 mg/mL sucrose, 10.7 mM sodium acetate, pH 7.5.
b This study was designed by the Sponsor and conducted by the University of Texas Medical Branch.
c mRNA-1647 contains 6 mRNAs which encode the full-length CMV gB and the pentameric gH/gL/UL128/UL130/UL131A glycoprotein complex. The 6 mRNAs are formulated at a target mass ratio of 1:1:1:1:1:1 in a mixture of 4 lipids (SM-102, PEG2000-DMG, cholesterol, and DSPC) and formulated in 93 mM Tris, 60 mM NaCl, and 7% PG.
d mRNA-1706 contains a single mRNA sequence that encodes the prME structural proteins of Zika virus combined in a mixture of 4 lipids (SM-102, PEG2000-DMG, cholesterol, and DSPC) and formulated in 20 mM Tris, 8% sucrose, pH 7.4.
e A Good-Laboratory Practice study.
f mRNA-1653 contains 2 distinct mRNA sequences that encode the full-length membrane-bound fusion proteins of hMPV and PIV3. The 2 mRNAs are formulated at a target mass ratio of 1:1 in a mixture of 4 lipids (SM-102, PEG2000-DMG, cholesterol, and DSPC) and formulated in 93 mM Tris, 7% PG, 1 mM DTPA, pH 7.4.
g mRNA-1893 contains a single mRNA sequence that encodes the prME structural proteins of Zika virus in a mixture of 4 lipids (SM-102, PEG2000-DMG, cholesterol, and DSPC) and formulated in 100 mM Tris, 7% PG, 1 mM DTPA, pH 7.5.
h mRNA-1443 contains a single mRNA sequence that encodes for a phosphorylation mutant of the CMV pp65 protein (i.e., deletion of amino acids 435-438) combined in a mixture of 4 lipids (SM-102, PEG2000-DMG, cholesterol, and DSPC) and formulated in 93 mM Tris, 60 mM NaCl, and 7% PG.
MODNCO : The original dose levels selected were 0, 10, 30, and 100 μg/dose, respectively (SoA issued on 16 Mar 2017). The calculated dose levels were revised based on the updated concentration reported for mRNA-1443 Lot No. MTDP17017 (SoA issued on 30 May 2017). The change in the reported mRNA content for mRNA-1443 was 4%.
i NPI luciferase mRNA is combined in a mixture of 4 lipids (SM-102, PEG2000-DMG, cholesterol, and DSPC) and formulated in 25 mM Tris, 123 g/L sucrose, 1 mM DTPA, pH 7.5.
j Enty added from MODNCO : m mRNA-1273 contains a single mRNA sequence that encodes the full-length SARS-CoV-2 S-2P combined in a mixture of 4 lipids (SM-102, PEG2000-DMG, cholesterol, and DSPC) and formulated in 20 mM Tris, 87 mg/mL sucrose, 17.5 mM sodium acetate, pH 7.5.
m Enty added from MODNCO: The original dose levels selected were 0, 10, 50, and 100 μg/dose, respectively (SoA issued on 11 October 2016). The calculated dose levels were revised based on the updated concentration reported for mRNA-1706 Lot No. MTDP16064 (SoA issued on 03 May 2017). The change in the reported mRNA content for mRNA-1706 was 29%.
n MODNCO : The original dose levels selected were 0, 10, 30, and 100 μg/dose, respectively (SoA issued on 16 Mar 2017). The calculated dose levels were revised based on the updated concentration reported for mRNA-1647 Lot No. MTDP17015 (SoA issued on 31 May 2017). The change in the reported mRNA content for mRNA-1647 was –11%.
Abbreviations additional from original :
hMPV human metapneumovirus
PIV3 parainfluenza virus type 3
LIP4 SM-102, PEG2000-DMG, cholesterol, and DSPC)
MODNCO Moderna nonclinical overview (152)
NPI Nascent Peptide Imaging (NPI)
PG Abbreviation undefined in original. Possibly PEG = polyethylene glycol?
SD Sprague Dawley
SoA Summary of Analysis (from MODNCO)
Additional footnotes from original
** Assumed that only SM-102 tested and not complete LNP formulation per statement in first paragraph p50/169. Also see description of this study on p50/169 “ in vitro mammalian cell micronucleus test” but note typo there referring to study number 9601567 instead of 9601568.
## This should probably indicate footnote i missing from the body of the table on p44/169
%% see undefined abbreviation PG above
$$ Reference to footnote “i“ appears erroneous and likely should be footnote h, see p48/169
10.1.1. Was the modRNA that “constitutes the vaccine” used in genotoxicity studies?
It was not.
N one of the six genotoxicity studies conducted by Moderna used “the modified mRNA […] that constitute[s] the vaccine,” namely mRNA 1273. One study involved a Zika modRNA, one a luciferase model modRNA, and four involved LNP components.
The one biodistribution study (B1) involved a CMV modRNA vaccine.
None of the six GLP repeat dose toxicology studies (T1-T6) involved mRNA 1273. Two Zika, two CMV and one hMPV / PIV3 modRNA vaccine were evaluated in these studies.
Only two safety related studies did involve mRNA 1273:
· One non-GLP study repeat dose study (T7),
· One reproductive and developmental study (R1).
10.1.2. Were the formulations of mRNA 1273 and non-candidate vaccines comparable in safety studies?
The EMA assessment of the Moderna vaccine (136) reveals several formulation differences between the CMV vaccine (mRNA 1637) and Moderna’s candidate mRNA 1273 used in the biodistribution study and one of the toxicology studies (study T5 Table 1 ). Footnotes a and c to Table 1 describe:
· Footnote c: “mRNA-1647 contains 6 mRNAs which encode the full-length CMV gB and the pentameric gH/gL/UL128/UL130/UL131A glycoprotein complex. The 6 mRNAs are formulated at a target mass ratio of 1:1:1:1:1:1 in a mixture of 4 lipids (SM-102, PEG2000-DMG, cholesterol, and DSPC) and formulated in 93 mM Tris, 60 mM NaCl, and 7% PG. “
· Footnote a: “mRNA-1273 contains a single mRNA sequence that encodes for the full-length SARS-CoV-2 S-2P combined in a mixture of 4 lipids (SM-102, PEG2000-DMG, cholesterol, and DSPC) and formulated in 20 mM Tris, 87 mg/mL sucrose, 10.7 mM sodium acetate, pH 7.5.“ This composition appears to be that of the development lots used in the nonclinical program described in Moderna’s nonclinical review (152) Error! Bookmark not defined. at a mRNA concentration of 0.5 mg/ml whose preparation was said to be comparable to the manufacturing process used to make in clinical trial material with a composition of mRNA 0.2 mg/mL in 20 mM Tris buffer containing 87 g/L sucrose and 4.3 mM acetate, at pH 7.5.
The differences ( Table 2 ) in Tris, sucrose, sodium acetate, NaCl and “PG” (see Question 210 ) content controvert FDA’s assertion that the test article in the biodistribution study was “manufactured using the same procedure as SPIKEVAX.” In the absence of any validating evidence to the contrary, these sorts of differences are likely to modulate various physicochemical properties of the LNPs (in addition to particle size) that could affect LNP distribution and transfection.
Table 2 : Comparison of formulations from Table 1
|
ID |
Vaccine |
LNP/ lipid |
Tris mM |
Sucrose |
OTH |
PG % |
pH |
Lot |
SoA |
Study # |
|||
|
Repeat-dose Toxicology Studies |
|||||||||||||
|
T1 |
Zika mRNA 1706 |
LIP4 |
20 |
8% |
7.4 |
MTDP16064 |
SoA 10/11/2016 SoA 03 May 2017 |
5002045 |
|||||
|
T2 |
Zika mRNA 1706 |
LIP4 |
20 |
8% |
7.4 |
5002231 |
|||||||
|
T3 |
hMPV / PIV3 mRNA 1653 |
LIP4 |
93 |
DTPA 1mM |
7 |
7.4 |
5002033 |
||||||
|
T4 |
Zika mRNA 1893 |
LIP4 |
100 |
DTPA 1mM |
7 |
7.5 |
5002400 |
||||||
|
T5 |
CMV mRNA1647 |
LIP4 |
93 |
NaCl 60mM |
7 |
MTDP17015 |
SoA 16 Mar 2017 SoA 31 May 2017 |
5002034 |
|||||
|
T6 |
CMV mRNA 1443 |
LIP4 |
93 |
NaCl 60mM |
7 |
MTDP17017 |
SoA 16 Mar 2017 SoA 30 May 2017. |
5002158 |
|||||
|
Other Toxicity Study |
|||||||||||||
|
T7 |
SARS-CoV-2 mRNA 1273 |
LIP4 |
20 |
8,7% |
Na Acetate 10.7mM |
7.5 |
2308-123 |
||||||
|
Biodistribution Study |
|||||||||||||
|
B1 |
CMV mRNA1647 |
LIP4 |
93 |
NaCl 60mM |
7 |
5002121 Amendment 1 |
|||||||
|
Genotoxicity Studies |
|||||||||||||
|
G1 |
None |
SM-102 |
bacterial reverse mutation |
9601567 |
|||||||||
|
G2 |
None |
SM-102 |
in vitro micronucleus |
9601568 |
|||||||||
|
G3 |
Zika mRNA 1706 |
LIP4 |
20 |
8% |
7.4 |
MTDP16064 |
In vivo micronucleus SoA 11 October 2016 SoA 03 May 2017 |
9800399 |
|||||
|
G4 |
Luciferase model mRNA |
LIP4 |
25 |
12.3 |
DTPA 1mM |
7.5 |
in vivo micronucleus assay |
AF87FU.125012 NGLPICH.BTL |
|||||
|
G5 |
None |
PEG2000-DMG |
bacterial reverse mutation test |
9601035 |
|||||||||
|
G6 |
None |
PEG2000-DMG |
in vitro micronucleus |
9601036 |
|||||||||
|
Reproductive and Developmental Toxicity |
|||||||||||||
|
R1 |
mRNA 1273 |
LIP4 |
20 |
8.7% |
Na Acetate 17.5mM |
7.5 |
20248897 |
||||||
10.1.3. Was LNP size comparable in Moderna’s safety studies?
The EMA document (136) notes (p47/169) that the “The amount of the LNPs in the test material differed slightly in particle size from the final vaccine formulation of mRNA-1273.”
This disclosure, omitted in the FDA document, (151) challenges FDA’s representation (p14/30) that because the test article was “manufactured using the same procedure as SPIKEVAX” it contained LNPs whose properties matched those of SPIKEVAX, including its “ biodistribution and retention” properties ( Figure 12 ) .
Moderna scientists published data showing that using a CMV vaccine in a model system, LNP particle size had a substantial difference on immunogenicity in mice but not in non-human primates. (153) The EMA document (p47/169 in (136) ) points out that the unknown impact of this size difference on mRNA distribution, although offers some speculation regarding the liver:
“Even though it is not straightforward to understand the impact that the different particle size might have on mRNA tissue distribution, if any, nevertheless the liver distribution is not affected because the average diameter of endothelial fenestrae in the liver sinusoids in the rats The observed biodistribution with smaller LNP particle size should thus represent a worst-case scenario .” (p47/169) (emphasis added)
Question 156: Given that, according to a paper co-authored by a founder of Moderna, (67) LNP particle size is a major determinant of distribution, and also according to FDA,“because biodistribution and retention is a property of the LNP rather than the mRNA,” how does this the study support“the approval of SPIKEVAX BLA”? Q156
Question 157: Following from Question 156, Given that, according to a paper co-authored by a founder of Moderna, (67) LNP particle size is a major determinant of distribution, and also according to FDA,“because biodistribution and retention is a property of the LNP rather than the mRNA,” how does this the study support the authorization of Pfizer product using Tris buffer? Q157
10.1.4. Other differences in manufacturing and formulation of mRNA 1647 used in the PK study
The study report of Moderna’s investigation, conducted by Charles River (131) provides evidence of further departure from the assumptions of formulation and manufacturing comparability between mRNA 1647 and mRNA 1273.
· The mRNA-1647 test article was supplied at a concentration of 1.9 mg/ml (p11/280) which required dilution. This differs from the 0.2 or 0.5 mg/ml concentration, requiring no dilution, depicted in the EMA (136) and (154) documents. How LNPs are stored and prepared would certainly have effects on their stability and physiochemical properties that could affect distribution.
· The diluent was phosphate buffered saline (PBS) at pH7.2 ( Figure 14 ).
See 8.2 for discussion of the effects of buffers.
|
|
|
|
p13/701 from (131) highlight added |
p14/701 (131) highlight added |


Figure 14 : Screenshots from Moderna’s distribution Study of mRNA-1647 in rats (131)
The analysis of the test article in Moderna’s report (131) has been redacted and therefore some details of the formulation are unknown, However, that PBS does not feature in any version of the Tris-based buffer described above controverts the premise of formulation and manufacturing comparability.
Question 158: Given that the composition of mRNA 1647 is critical to understand the relevance of any studies that are used to support the authorization or approval of mRNA 1273, please provide the full formulation details of mRNA 1647, such as those redacted from the distribution study report.(131) Q158
Question 159: Per Question 158, did the formulation of mRNA 1647 used in Moderna’s distribution study contain Tris? Q159
Question 160: Given the manufacturing controls alluded to by Moderna (129) to reduce lipid adduct formation, when, relative to the conduct of the Moderna’s distribution study, were these controls implemented? Q160
Question 161: Please confirm the accuracy of FDA’s document(151) in stating the that the product used in Moderna’s distribution study was “manufactured using the same procedure as SPIKEVAX”? Q161
10.1.5. Inadequate preclinical use of animals with spike-ACE2R binding relevant characteristics
An additional concern relates to the choice of animal models for nonclinical studies. Efforts have been made to identify or develop animal models to study the pathophysiology of SARS-Cov-2 infection or to develop therapeutics. (155) A particular challenge has been to model the binding of the viral spike protein which mediates cell entry, particularly its interactions with the ACE2 receptor. Jiang et al. noted: (156)
“The predicted 29 amino acid residues of ACE2 that interact with SARS-CoV-2 spike protein receptor binding domain (RBD) vary between human ACE2 and mouse or rat ACE2. Therefore, wildtype mice and rats show lower SARS-CoV-2 infection rate and mild symptoms compared to what is seen in humans. Small animal models that recapitulate human COVID-19 disease are urgently needed for better understanding the transmission and therapeutic measurement. “
Brooke and Prischi (157) conducted structural and functional modeling studies in several species with the SARS-Cov-2 spike protein, and the three main proteins with which it acts: ACE2 receptor, TMPRSS2 and furin. While TMPRSS2 and furin are largely conserved between species, the sequence of ACE2 is more variable, influencing spike protein binding. Of the species studied, macaque, ferrets and hamster appeared most suitable to study the effects of anti-SARSCov-2 antibodies and small molecule inhibitors of the spike-ACE2 interaction. Rats, along with rabbits, Guinea pigs, mice, were determined not to constitute a suitable model. The authors suggested that transgenic animals expressing human ACE2 would likely provide useful for models of viral entry. Both mouse and rat models expressing human ACE2 have been developed. (156) Although the murine version is used more commonly, (e.g. (158, 159) ) a rat hACE2 model is also commercially available. [23]
In selecting the macaque for immunogenicity and viral challenge studies, Pfizer (160) acknowledged the importance of an ACE2-relevant species:
“The human and rhesus ACE-2 receptor have 100% amino acid identity at the critical binding residues, which may account for the fidelity of this SARS-CoV-2 animal model.” (p12/36)
The challenge of selecting an appropriate animal model was acknowledged by Moderna in the context of immunogenicity studies:
“Wild-type (WT) mice are a convenient and easy-to-use model to assess vaccine immunogenicity; however, the ACE-2 receptor, the primary route for SARS-CoV-2 binding and entry, differs significantly between mice and humans and, as result, WT SARS-CoV-2 does not infect mice.” (p7/31) (152)
Moderna continues and explains that its approach in this case was to use a mouse-adapted SARS-Cov-2 strain:
“Therefore, a mouse-adapted SARS-CoV-2 strain, which was developed by the laboratory of Dr. Ralph Baric at the University of North Carolina at Chapel Hill, was used to assess protection of immunized mice from SARS-CoV-2 challenge. “(p7/31) (152)
Moderna also discussed the related question of selecting suitable animal models to study vaccine-associated ERD. “ (p7/31) (152)
Clearly both Pfizer and Moderna understood the importance of ACE2-relevant models for various aspects of their nonclinical programs. However. since some toxic or genotoxic effects of COVID-19 mRNA vaccines may be related to the distribution of expressed spike protein, likely dependent in part on ACE2 binding, similar considerations of an ACE2-relevant model should also apply to t oxicity and genotoxicity studies, as well as to studies of spike protein expression.
This was not the case, as the rat was chosen based, at least according to Moderna, (p8/31) (152) because of its acceptance as a species for nonclinical toxicology testing by regulatory agencies.
Question 162: What consideration has FDA made concerning the selection of suitable animal models for pharmacology, biodistribution and other safety studies for modRNA products encoding antigens whose interaction with host ligands may be species specific? Q162
11. Inadequate studies failed to predict modRNA and spike protein PK and expression kinetics
When asked about the kinetics of spike protein production at the June 15 2022 VRBPAC meeting, [24] the Pfizer representative dismissed the question as “somewhat academic. ” We disagree. Central to understanding the pharmacology and toxicology of the modRNA vaccines is a consideration of
· the biodistribution [25] of the vaccines and their components (including residual DNA)
· spike protein expression and distribution kinetics.
· transfection of the LNP and mRNA release into the cytosol
The terms “biodistribution” and “transfection” must be clearly distinguished from “gene expression.” (161) Biodistribution refers to the physical location of a drug, tracer, or intact LNP within a biological system. It depends on circulation, the protein corona, vascular permeability, and reticular endothelial system (RES) uptake but does not indicate cell entry. Transfection is the process of delivering nucleic acids, such as modRNA. It requires both cellular uptake and endosomal release. In contrast, gene expression involves translating the modRNA into the target protein, such as the SARS-CoV-2 spike protein. This process depends on the intact modRNA, active ribosomes, and protection from degradation. It is important to recognize that the inherent instability of LNPs, combined with degradation, opsonization, and various biological barriers, can cause modRNA shearing, premature release, or even LNP disassembly, which can significantly reduce their therapeutic effectiveness. (162)
11.1. Discrepancies between messaging and data on modRNA, spike persistence and distribution
As discussed earlier ( 4.3.1 ), public health agencies have argued that “the mRNA is destroyed.” This is reflected in the messaging behind CDC’s vaccination campaign, shown in this screen shot ( Figure 15 ) from the CDC web site [26]

Figure 15 : Screen shot from CDC web site: “do not last long”
This page represents that the vaccinal modRNA is eliminated within a few days, and the spike protein within a few weeks after vaccination. This information is inconsistent with emerging reports that extend these estimates to weeks or months for both modRNA (72, 73, 75) and spike protein (74, 75, 163) (see also review in (78) ).
Another point of variance between public health messaging and available data is that modRNA distributes far more widely than represented. This screen shot from the same CDC web page fails to disclose the wide distribution known from animal studies conducted by Pfizer (160, 164, 165) and Moderna (136) , conveying the impression that the vaccine “stays in the arm muscle.”

Figure 16 : Screen shot from CDC web site: enters muscle cells (highlight added)
Although much was known about (164, 165) about the distribution of LNPs, the discrepancy between what was represented and what is now known appears was exacerbated due to the non-conduct of adequately informative pharmacokinetic studies. Limited non-clinical studies were performed with vaccine prototypes whose composition differed from the candidate COVID-19 modRNA vaccines. No clinical pharmacokinetics studies appear to have been performed, based on the content of sponsor and FDA VRBPAC briefing documents or review memoranda for Pfizer (166-168) and Moderna. (169-171)
The non-conduct of critical studies appears to have been justified by:
· unsupported expectations about modRNA, protein production (or gene expression) and LNP kinetics
· a failure to follow guideline provisions for case-by-case consideration
· a superficial analysis of the limited studies that were performed, lacking follow up
· An inappropriate reading of WHO guidelines (in Pfizer’s non-clinical overview (160) that “Pharmacokinetic studies have not been conducted with BNT162b2 and are generally not considered necessary to support the development and licensure of vaccine products for infectious diseases (WHO, 2005; WHO, 2014).””
11.2. Unsupported expectations about modRNA, protein expression and LNP kinetics justify study non-conduct
“Expectations” on the part of sponsors and/or regulators about the metabolism and kinetics of nucleoside modified RNA, spike protein production and LNPs were used to justify the non-conduct of critical PK and expression kinetics studies.
11.2.1. Unsupported expectations about modRNA and protein metabolism
Pfizer’s non-clinical overview from February 2021 released under FOIA states (p20/36): “The protein encoded by the RNA in BNT162b2 is expected to be proteolytically degraded like other endogenous proteins. RNA is degraded by cellular RNases and subjected to nucleic acid metabolism. Nucleotide metabolism occurs continuously within the cell, with the nucleoside being degraded to waste products and excreted or recycled for nucleotide synthesis. Therefore, no RNA or protein metabolism or excretion studies will be conducted .” (160) (emphasis added)
No data appear to have been cited to support justify these expectations.
Question 163: What data or literature were provided by Pfizer to support their expectations (160) regarding the degradation or mRNA or spike protein? Q163
Pfizer’s expectations are mirrored by WHO’s similarly unsupported contention in their 2021 guideline on mRNA vaccines: (71)
“the vaccine mRNA degrades within a relatively short time once taken up by the body’s cells, as does the cell’s own mRNA. During that entire time, the mRNA vaccine is expected to remain in the cytoplasm, where it will be translated and then degraded by normal cellular mechanisms.” (p48/66) (emphasis added)
The drafting of this document, like others to be cited shortly, (48, 172) has enjoyed FDA’s acknowledged participation .
Question 164: Given emerging data suggesting vaccinal modRNA persistence for significantly longer (72, 73, 75, 78) than the “short time” described in the WHO guideline on mRNA vaccines,(71) and given FDA’s participation in the drafting of that document, what revisions has FDA proposed or will propose to that document? Q164
Question 165: Since the introduction of the modRNA COVID-19 vaccines, to what extent did FDA agree with CDC messaging suggesting that the modRNA is eliminated “within a few days” and the spike protein “within a few weeks” (see 11.1))? Q165
Question 166: Given emerging data suggesting vaccinal modRNA persistence for significantly longer (72, 73, 75, 78) than the “few days” and spike protein persistence for significantly longer than “a few weeks”(74, 75, 78, 163) (see 11.1) what revisions has FDA proposed or will propose to CDC or other government entities to correct the earlier statements? Q166
11.2.2. Unsupported expectations about local distribution and action
Another expectation supporting the non-conduct of pharmacokinetic studies concerns the notion that vaccines or their adjuvants remain or act locally, found in the following WHO guidelines. (172) (48)
The 2014 WHO guidelines on adjuvanted vaccines (172) state with some qualification: “Adjuvants are expected to exert their action locally in close connection to the antigen. However, biodistribution studies can be helpful in understanding the distribution of the adjuvant following injection” (p26/42) (emphasis added).
The 2021 WHO guideline on DNA plasmid vaccines (48) discounts the need for distribution studies: “Although biodistribution studies were previously suggested for DNA vaccines, the data acquired to date have not shown reason to continue with such evaluations. Plasmid DNA remains largely at the injection site and does not biodistribute at clinically relevant levels or widely throughout the body . Furthermore, it does not target the ovaries or testes and clears from the body by degradation.” ( p32/54) (emphasis added, citations omitted)
These expectations are inconsistent with animal data known to regulators (164, 165) but appear consistent with public health messaging suggesting that the modRNA vaccines stay at the site of injection, where they act (see 11.1 ).
Question 167: Since the introduction of the modRNA COVID-19 vaccines and given data generated by Pfizer and Moderna in animals showing a wide distribution of LNPs and/or modRNA, to what extent did FDA agree with CDC messaging suggesting that the modRNA vaccines stay at the site of injection, where they act (see 11.1)? Q167
Question 168: To what extent does FDA now agree with CDC messaging suggesting that the modRNA vaccines stay at the site of injection, where they act (see 11.1)? Q168
Question 169: Given Pfizer’s and Moderna’s data from animals showing a wide distribution of LNPs and/or modRNA and given FDA’s participation in the drafting of WHO guidelines (48, 172) suggesting a much narrower distribution, what revisions has FDA proposed or will propose to those document regarding vaccines if any kind that use LNP-technology? Q169
11.2.3. Unsupported expectation about LNP and mRNA kinetics
Limited non-clinical PK studies were performed with modRNA vaccine prototypes whose compositions differed from the candidate COVID-19 modRNA vaccines for which no studies were reported.
The principal difference between the tested and candidate vaccines was in the sequence of the modRNA payload carried by the LNPs. Other differences are largely unknown in terms of method of manufacture, nucleoside modification, codon optimization, amounts and types of impurities and contaminants. Small differences in LNP composition, physicochemical attributes or manufacturer may impact a number of properties, including distribution. (67)
A major goal in conducting PK studies is to characterize the distribution, production, and persistence of the spike protein, as these factors presumably relate to the extent and type of immune response it will trigger. Spike PK properties depend on modRNA PK and expression characteristics, which in turn depend on LNP PK properties. Understanding the PK and expression properties of the spike protein, modRNA, and LNPs is essential, as discussed by Vervaeke et al. (173) Additionally, the PK properties of residual DNA or other impurities must be understood to inform about potential risks (see Question 86 ).
Aside from the tenuous expectations as to spike or modRNA persistence and distribution,( 11.2.1 ) localization ( 11.2.2 ) or generous interpretation of guidelines, ( 11.3 ) another reason why regulators have accepted PK studies involving LNP vaccines carrying non-candidate modRNA is, as expressed in FDA’s “Summary Basis for Regulatory Action” for SPIKEVAX, that “Because biodistribution and retention is a property of the LNP rather than the mRNA, results from this study were considered supportive for the approval of SPIKEVAX BLA.” (p14/30) (151)
FDA’s statement reaches further than did Moderna’s nonclinical review: “ The distribution, toxicity, and genotoxicity associated with mRNA vaccines formulated in LNPs are driven primarily by the composition of the LNPs and, to a lesser extent, by the biologic activity of the antigen(s) encoded by the mRNA.” (p p 316 and 676 in (174) ) Likewise EMA’s assessment report (136) did not paint the same picture as FDA of distribution of mRNA being the sole property of the LNP:
“It is biologically plausible that the distribution of the mRNA vaccine is determined by the lipid nanoparticle content, whereas the influence of the mRNA itself is considered very limited. Therefore, it is acceptable that the biodistribution study was performed with the same lipid nanoparticles containing another mRNA (i.e. mRNA-1647).“ (p47/169)
This premise, underlying a platform approach, is open to significant challenge.
· LNPs constitute a delivery method for modRNA, much like a buffer solution in a hypodermic syringe is a system for delivering drugs. The initial phases of distribution must certainly be highly dependent on the properties of the delivery system (physicochemical properties such as particle size, payload, composition etc. for LNPs, buffer composition needle size and administration rate for the syringe). The general chemistry of mRNA will likely determine to a large degree its general distributive properties. However, a close inspection of Moderna’s study reveals that although PK parameters for six different CMV mRNAs (131) delivered in the same LNPs were mostly (but not always) similar at the injection site and neighboring lymph nodes. up to 12-fold differences in key pharmacokinetic parameters between the different mRNA constructs in other tissues. (see 11.4.2 ).
The assertion that “ biodistribution and retention is a property of the LNP rather than the mRNA” is further misleading because it fails to address how pattern of protein expression may differ with different mRNA constructs, in different formulations.
· That the distribution of mRNA delivered by LNP may be partly dependent on the sequence of its modRNA payload is suggested by work conducted by BioNTech in which the distribution of modRNA delivered by liposomes was dependent on the modRNA sequence, particularly that of the 3’ UTR (175)
· RNA stability is dependent on a number of features such as splicing patterns, transcript length, and G+C content, (176) not to mention of course nucleoside modification with N1 methylpseudouridine.
· The distribution of the encoded protein will depend on its own biochemistry. A recent review (78) pointed out how results from Pfizer’s studies ( 11.5 ) on the kinetics of luciferase mRNA expression measured by bioluminescence would not model the distribution of spike protein which is a transmembrane protein that can circulate within episomes. Furthermore, the duration of luciferase expression may not be representative of the duration of spike protein expression
The validity of this premise not only affects the applicability of PK studies conducted with non-candidate versions of modRNA vaccines, it affects the relevance of most other non-clinical studies where these materials are used as test articles. These submissions only provide (albeit inadequate) biodistribution data and lack information on transfection or gene expression, which does not accurately reflect the true nature of these modRNA-LNP vaccines.
Question 170: What data has FDA relied upon to validate the assertion that “biodistribution and retention are properties of the LNP rather than the mRNA”? Q170
Question 171: Why has the FDA not required data on the biodistribution of the spike protein? That is, have cells been transfected and have subsequently produced the desired protein? Q171
11.3. Failure to follow guideline provision for case-by-case consideration
In Pfizer’s non-clinical overview (160) the non-conduct of pharmacokinetic studies on the BNT162b2 candidate was justified because they “are generally not considered necessary to support the development and licensure of vaccine products for infectious diseases,” (p17/36) citing guideline documents from WHO issued in 2005 (177) and 2014. (172) A closer inspection of these two documents reveals how the term “ generally not considered necessary” has been generously construed.
The first of these, the 2005 document (177) addressing vaccines points to the need to consider these sorts of studies on a case-by-case basis when new formulations or adjuvants are involved:
“Pharmacokinetic studies (e.g. for determining serum or tissue concentrations of vaccine components) are normally not needed . The need for specific studies should be considered on a case-by-case basis (e.g. when using novel adjuvants or alternative routes of administration) and may include local deposition studies that would assess the retention of the vaccine component at the site of injection and its further distribution (e.g. to the draining lymph nodes). Distribution studies should be considered in the case of new formulations , novel adjuvants or when alternative routes of administration are intended to be used (e.g. oral or intranasal).” (p21/36) (emphasis added)
The second, 2014, document addressing vaccine adjuvants and adjuvanted vaccines (172) also points to a case-by-case consideration: “The feasibility of and need for such biodistribution studies should be evaluated on a case-by-case basis.” (p26/42) (emphasis added)
The guidelines (172, 177) cited by Pfizer (160) to justify the non-conduct of pharmacokinetic studies because they “are generally not considered necessary” contemplate traditional vaccines, with the typical “vaccine component” consisting of the target antigen or killed or attenuated target organism. These guidelines do not contemplate modRNA vaccines that elicit production of the ultimate antigen in the body of the vaccinee. These guidelines as well as those for adjuvants (172) and plasmid vaccines (48) do not contemplate delivery by a novel LNP formulation whose distribution (see below) clearly extends beyond the injection site.
Even if the WHO guidelines (172, 177) could apply to modRNA vaccines rather than provide the implied blanket exemption for conducting pharmacokinetic studies of modRNA or spike antigen, both documents clearly speak of the need to consider these questions on a case-by-case basis, given the vaccines’ novelty. This has been circumvented by invoking “ generally not considered necessary.” (emphasis added)
Regarding the kinetics of spike protein production, the use of this blanket exemption is all the more puzzling given that the WHO 2007 plasmid DNA guidelines (49) acknowledges: “ Knowledge of the duration of expression of an antigen from injected DNA is limited although some reports suggest that expression could continue for many months , which means that the possibility of tolerance may remain a concern. (p19/25) (emphasis added)
Question 172: Given the novel mechanism of action, delivery and distribution of modRNA vaccines is not contemplated by WHO guidelines (172, 177), please provide a rationale for why they can be used to justify the non-conduct of RNA or protein metabolism or excretion studies on the candidate vaccine formulations? Q172
11.4. Superficial analysis of limited biodistribution studies, lacking follow-up: Moderna
FDA’s summary document for SPIKEVAX cited by Dr. Marks (151) (p14/30) notes the performance of a biodistribution study in rats of a vaccine of redacted identity. This study appears to correspond to study B1 ( Table 1 ) described in the EMA document (136) on a similarly formulated CMV mRNA (mRNA 1647) vaccine containing 6 mRNAs, whose levels were measured directly. The EMA alluded (p47-48/169) to WHO guidelines on the general acceptability of not needing pharmacokinetic studies for vaccines (see 11.3 ) and acknowledged the need for distribution studies “in the case of new formulations or novel excipients used.” [27]
Several documents describe Moderna’s biodistribution study, with sometimes apparently conflicting details.
· The study report itself as amended. (131)
· FDA Summary Basis for Regulatory Action, as you cite (151) Jan 30 2022
· EMA Assessment report (136) Mar 11 2021
· Moderna Nonclinical overview. (152) Three versions of this document have been released under FOIA.
· Moderna Pharmacokinetics Written Summary. (154)
· Moderna Pharmacokinetics Tabulated Summary. (178)
An examination of these other documents reveals details omitted from the cited FDA document that:
· affect an assessment of the study’s quality and its results.
· challenge FDA’s representation that mRNA 1647 (p14/30) of (151) was “manufactured using the same procedure as SPIKEVAX” implying that other than the mRNA sequence, the formulation of the test article was otherwise identical to mRNA-1273.
· challenge the premise that “because biodistribution and retention is a property of the LNP rather than the mRNA,” the study supports “ the approval of SPIKEVAX BLA.”
· challenge the expectations regarding rapid elimination of modRNA and narrow distribution.
11.4.1. Quality and methodology issues
· The non-GLP status and non-inclusion of female rats do not qualify this study as pivotal, according to EMA. (p52/169 in (136) )
· The longest time point in the study was 120 hours after a single dose.
· Levels of mRNA were assessed using a multiplex branched DNA (bDNA) assay. A footnote in Moderna’s Pharmacokinetics Tabulated Summary. (178) states that the “ method was not formally validated.” Other documents refer to this assay as a “Qualified bDNA method ” (eg p132/280 in (131) ), also (154, 178)
Question 173: What studies were conducted to establish that the biodistribution of modRNA incorporated into formulations used in the mRNA 1647 and mRNA 1273 test articles in Moderna’s toxicology and biodistribution studies, is equivalent? Q173
Question 174: How does the distribution and gene expression of mRNA as lipid-adduct compare with that of non-adducted mRNA? Q174
Question 175: Is mRNA as lipid-adduct detected in the multiplex branched DNA (bDNA) assay used to determine levels of mRNA in tissues in Moderna’s biodistribution study? Q175
Question 176: Does the presence of mRNA as lipid-adduct confound in any way the results and interpretation of Moderna’s biodistribution study and its gene expression? Q176
In their paper, (128) Moderna’s scientists stated that the lipid-mRNA adducts “render the mRNA untranslatable, leading to loss of protein expression.” If one of the primary reasons for conducting a biodistribution study is to understand how and where in the body protein expression takes place, any characterization of distribution that is confounded by the presence of mRNA-lipid adducts is of extremely limited value. This has implications for any toxicology study performed on prototypes where mRNA-lipid adducts may have been involved. (see 12.3.4 ) Further, in the Fall of 2021, Pfizer’s implemented a change in the buffer of their COVID-19 mRNA vaccine from PBS to Tris. (see 8.2 ).
Question 177: Given thattheformulationof the mRNA 1647 used in Moderna’s toxicology and biodistribution studies appear to differ substantially from mRNA 1273 in ways that likely materially affect LNP physicochemical, distribution and transfection properties, how do studies involving mRNA 1647 support “the approval of SPIKEVAX BLA”? Q177
11.4.2. Evidence for mRNA dependent distribution
mRNA was found (p47/169 in (136) ) at the muscle of the injection site, proximal and distal lymph nodes, spleen and, except the kidney, all examined tissues, including heart, lung, testis, and brain tissues. Although there is rapid clearance from the plasma in the first 24 hours with a t1/2 of 2.7 - 3.8 hours, the half-lives for clearance from injection site muscle, proximal and distal lymph nodes, and spleen for all modRNA vaccine components were 14.9, 34.8, 31.1 and 63.0 hours, respectively. This means that the maximum time for 90% elimination could be as long as approximately 9 days, already somewhat longer than “ a few days .” ( 11.1 )
Moderna’s pharmacokinetic summary stated that the “ biodistribution of mRNA-based vaccines in LNPs is predicted to be driven by the LNP characteristics.” (p12/13 in (154) ). Six different CMV mRNA constructs studied were said to exhibit “nearly identical pharmacokinetic behavior.” (p11/701) (131) Although, a relatively small fraction of the administered mRNA-1647 dose distributed to distant tissues, with generally similar kinetics, a closer examination of the PK data revealed up to 12 fold differences in key pharmacokinetic parameters between the different mRNA constructs for different organs ( Table 3 ).
Table 3 reproduces Table 4 from Moderna’s report (p298/701) with green and pink highlights added to indicate examples of the wide range of values between mRNA constructs, for one or more parameters, for most of the tissues examined.
Table 3 : Table 4 from Moderna’s: Tissue Pharmacokinetic Parameters for a Single IM Dose of 100 μg of mRNA-1647 in Male Sprague Dawley Rats (154) (highlighted per text)
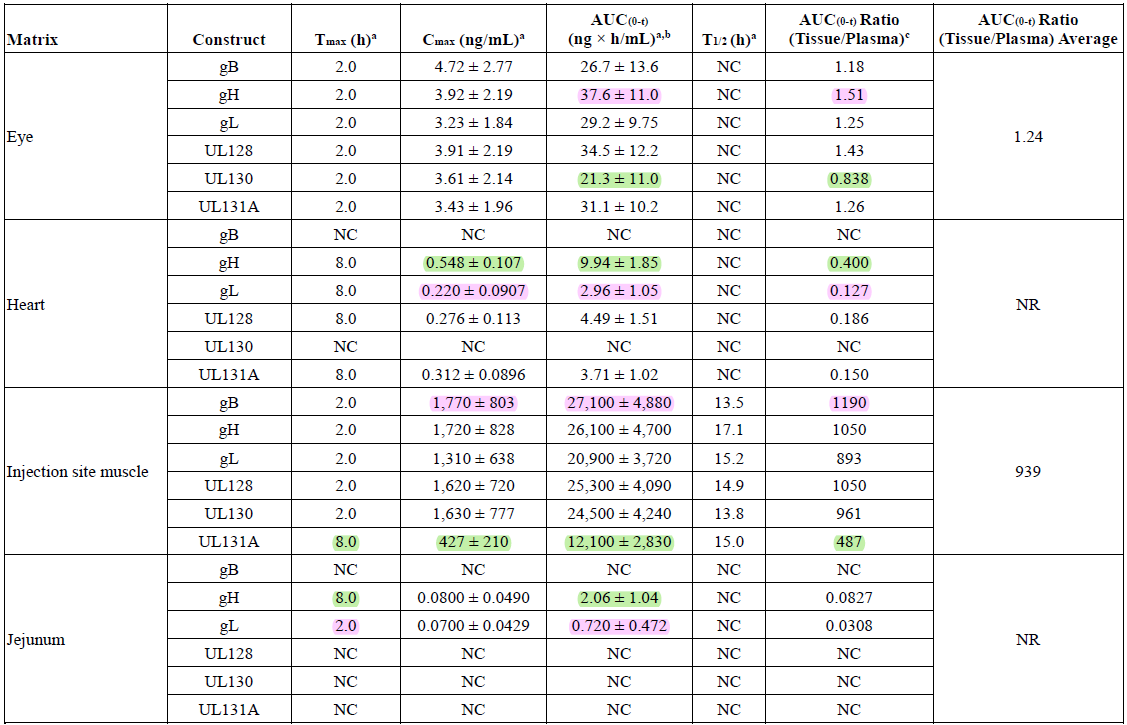
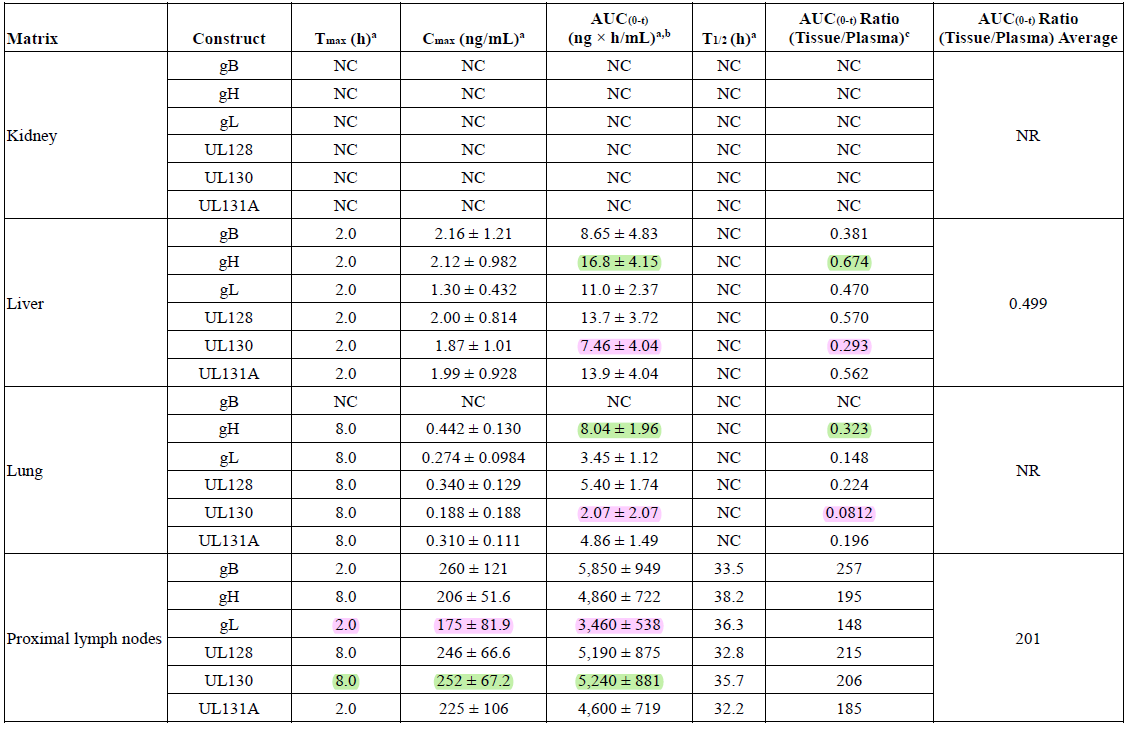
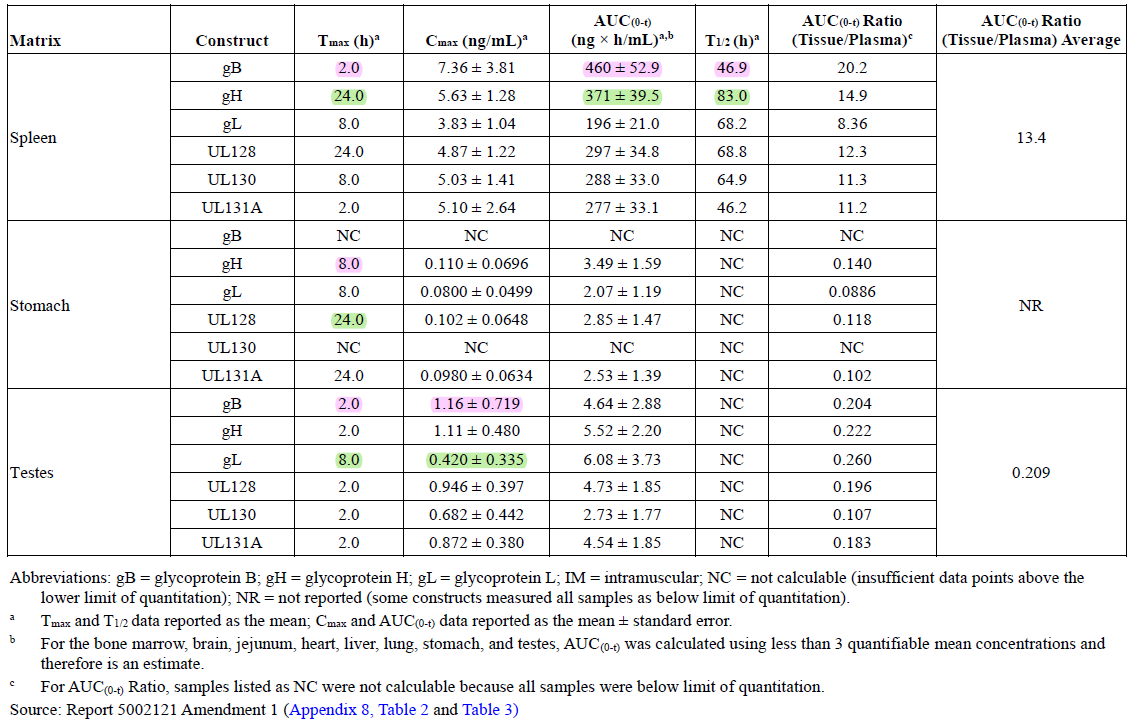
These data constitute an unintended experiment that effectively controls for between-LNP and between-animal variation by loading the six mRNA constructs in the same LNPs, [28] and injected into the same animals.
PK parameters for the six constructs at the injection site and the proximal and distal lymph nodes are mostly (not entirely, see Table 3 ) clustered tightly, perhaps unsurprisingly. In terms of the half-life of elimination from the injection site, the values for the six mRNA constructs cluster around 15 hours. For the proximal lymph nodes, they cluster around 35 hours. Accordingly, the time for 90% elimination is approximately 2-5 days, corresponding to CDC’s “few days” ( 11.1 ). Outside of these tissues there appears to be greater between-construct variability in PK parameters. In the spleen, the half-life is longer for all constructs, up to 83 hours for the gH construct, corresponding to a time to 90% elimination of approximately over 11 days.
Question 178: Per Question 170, and in view of the findings in Moderna’s own PK study suggesting construct-dependent kinetics, controverting FDA’s premise of almost exclusively LNP-dependent kinetics, what studies will FDA soliciting to better characterize the PK of the Moderna COVID-19 mRNA vaccine? Q178
Question 179: In view of the findings in Moderna’s own PK study suggesting construct-dependent kinetics, controverting FDA’s premise of almost exclusively LNP-dependent kinetics, what guidance will FDA issue regarding the sorts of PK studies needed to support mRNA product approval? Q179
These findings suggesting construct-dependent kinetics have implications for the reliance on toxicology studies performed with constructs unrelated to the candidate product to support licensure. If kinetics of mRNA constructs differing in sequence are even somewhat dependent on sequence, toxicology findings drawn from non-candidate constructs cannot be said to be representative of what might occur with the candidate construct.
Question 180: In view of the findings in Moderna’s own PK study suggesting construct-dependent kinetics, controverting FDA’s premise of almost exclusively LNP-dependent kinetics, what assurance can FDA give about the safety of a product whose approval has relied exclusively on toxicology studies conducted with non-candidate constructs? Q180
These studies were conducted to evaluate the toxicity and distribution of the LNPs alone, as they were considered novel excipients. (136) . Excipients are generally believed to have no physiological effect by themselves, since they function as carriers for the modRNA. Regulatory assessment of excipients in medicinal products (179) does not require a separate review process, such as pharmacokinetic (PK) studies. No biodistribution studies using the actual modRNA construct from the Pfizer/BioNTech or Moderna vaccine were included in the regulatory documents. As a result, there was no assessment of transfection efficiency or gene expression levels. This aligns with studies required for LNPs as excipients. Further clarification from regulatory authorities and manufacturers is needed to determine the necessary chemical, pharmacological, and toxicological studies for these lipids to obtain approval. (180) LNPs, or LNP components may in fact have adjuvant activity, as indicated by a 2022 FDA presentation, slide 20 of which states “Ionizable cationic lipid […] Provides adjuvant activity to LNPs” (181) Possessing adjuvant activity would trigger higher regulatory scrutiny. (172, 182)
11.5. Superficial analysis of limited biodistribution studies, lacking follow-up: Pfizer
In addition to studies on the lipid components, Pfizer conducted three limited biodistribution studies [29] on the distribution of LNPs or the expression kinetics of a model payload - mRNA encoding for luciferase. The two studies involving IM injection are of most interest.
· Study 18530 (164) of the distribution of radiolabeled LNPs in rats is extremely limited because it proceeded only for 48 hours, using a different mRNA (luciferase) and made by a process with unknown similarity to that used for commercial scale. The type of nucleoside modification, codon optimization and untranslated sequences are unknown. The study did not examine luciferase expression. The findings of LNP accumulation in a number of tissues, including, adrenal glands, bone marrow, liver, lymph nodes, ovaries, spleen, and to a smaller extent, testes, are concerning and controvert the expectation of localization expressed in the 2014 WHO document, (172) and should have triggered follow up investigation.
· The same concerns about mRNA composition and manufacture apply to study R-20-0072 (p17/36 in (160) ) of similarly loaded LNPs in mice. Unlike the previous study mRNA expression was examined, although the bioluminescence whole-body method is extremely crude showing distribution mainly to the liver and injection site. Nonetheless, the signal for luciferase expression remains above the control level even at 9 days ( Figure 17 ).
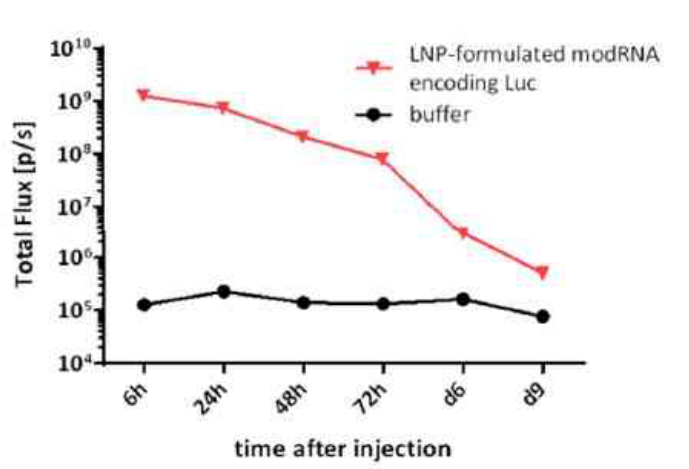
Figure 17 : Pfizer luciferase expression -PK study From figure 2.4.3-2 in study R-20-0072 (p17/36 in (160) ),
11.6. Biodistribution studies: summary
It is evident that these studies are woefully inadequate to characterize and deconvolute the kinetics of LNP biodistribution, transfection, modRNA persistence, protein expression, and persistence. According to Pfizer’s document (160) RNA or protein metabolism studies were not conducted. Moderna allude to in vivo expression studies (p7/31 in (152) ).
Question 181: What in vivo studies did Pfizer or Moderna provide to describe the distribution and kinetics of spike protein production after dosing with COVID-19 modRNA vaccines? Q181
A full understanding of the pharmacokinetics of any drug is of course essential to understand its pharmacology and toxicology. This is not an academic nicety. The biodistribution studies supporting the authorization and approval of the Pfizer and Moderna COVID-19 modRNA vaccines were inadequate for a variety of reasons. This inadequacy detracts from a full understanding of the pharmacology and toxicology impeding a comprehensive assessment of risks and benefits.
Question 182: Given reports of vaccinal modRNA or spike protein persistence far longer than indicated by Pfizer’s limited PK data or public health messaging, what animal or human studies have FDA requested of Pfizer and Moderna to better understand the PK of COVID-19 modRNA vaccines and to better inform a risk benefit analysis? Do these include studies using commercially available product? Q182
Question 183: Given reports of vaccinal modRNA or spike protein persistence far longer than indicated by Pfizer’s limited PK data or public health messaging, what guidance will FDA provide regarding the types of animal and human PK, distribution and expression kinetics studies should be performed for modRNA vaccines or other modRNA gene therapies? Q183
The concerns about lipid adducts and the actual formulations used in these studies as well as the assumptions about exclusively LNP dependent distribution has implications for the interpretation of the toxicity and genotoxicity studies since adducts decrease spike protein production as they are non-functional yet have intrinsic toxicological effects (see 12.3.3 12.3.3 ).
12. Safety studies: preclinical genotoxicity
Responding to concerns about genomic integration of residual DNA into the genome, mutagenesis, and cancer risk. Dr. Marks made these two statements regarding the results of genotoxicity studies conducted in animals.
· “Additionally, animal studies with the mRNA delivery technology done over the past decade show no evidence of genotoxicity.”
· “Additionally, studies have been conducted in animals using the modified mRNA and lipid nanoparticle together that constitute the vaccine, including the minute quantities of residual DNA fragments left over after DNAse treatment during manufacturing, and demonstrate no evidence for genotoxicity from the vaccine .” Footnote 3 to this statement contains two URLs, pointing to the “Summary Basis for Regulatory Action” documents for COMIRNATY (47) and SPIKEVAX. (151)
In summary, this response provides no evidence of any directly relevant genotoxicity studies being performed and is therefore wholly inadequate for the following reasons:
· The document cited regarding COMIRNATY (47) contains no reference whatsoever to animal genotoxicity studies.
· The document cited regarding SPIKEVAX (151) contains a reference to a rat micronucleus assay to evaluate “the genotoxic potential of (b) (4) [redacted] mRNA in SM-102 LNP” that “revealed no genotoxic effects of SM-102 LNP.” The identity of the mRNA studied is redacted, but EMA (136) and Moderna documents (152) describe micronucleus assays being performed on two mRNA vaccines – a luciferase model vaccine and ZIka virus. Several points emerge:
o These studies were NOT performed as represented using “ the modified mRNA and lipid nanoparticle together that constitute the vaccine.”
o The pattern of distribution, and therefore toxicity after IV the dosing used in these studies may differ from the IM dosing used clinically. O nly single doses were tested.
o The EMA (136) and Moderna documents (152) described no evidence of genotoxicity from the luciferase study, consistent with the FDA document. (151) .
o The Zika mRNA vaccine study did, according to both Moderna and EMA, yield positive findings of statistically significant increases in micronucleated erythrocytes in both sexes. The possible validity of mitigating circumstances discussed by EMA that might account for these observations, does not detract from the fact that its absence from the body of evidence referenced by Dr. Marks (151) to allay concerns serves only to heighten them and compounds reservations as to the integrity of regulatory process.
o Because assumptions that distribution is almost exclusively LNP dependent and largely independent of mRNA sequence are at best tenuous ( 11.4.2 ), reliance on favorable toxicological and genotoxicity findings is not firmly grounded.
· The rat micronucleus assay described in the cited SPIKEVAX document (151) and used to test the mystery mRNA is not an appropriate method of assessing risks of integration and mutagenicity, or, when used alone, carcinogenicity. Thus, even tests performed on the actual COVID-19 mRNA vaccine compositions would not address this concern.
· Other documents (e.g. 12.2 ) indicate that carcinogenicity (COMIRMATY, SPIKEVAX), mutagenicity (SPIKEVAX) or genotoxicity (COMIRMATY) were not performed.
· Any toxic or genotoxic effects of COVID-19 mRNA vaccines may potentially be related to the expressed spike protein. The distribution and kinetics of protein expression is therefore important to understand. As the spike protein is known to interact with the ACE-2 receptor, distribution, thus the use of animals in toxicity or genotoxicity whose ACE-2 is similar to that of a human is vital.
· There is no discussion of the lenient interpretation of WHO guidelines concerning the conduct of genotoxicity or carcinogenicity studies for vaccines (see 12.4 ).
· FDA guidance on the factors to consider for carcinogenicity studies of pharmaceuticals have been discounted (see 12.1 )
Before elaborating on these points, a brief review of the types of tests available and their relevance is worthwhile.
12.1. What kinds of tests are appropriate to assess integration, mutagenicity or carcinogenicity risk?
FDA has defined genotoxicity, also called “g enetic toxicity” (183) as: “ A broad term that refers to any deleterious change in the genetic material regardless of the mechanism by which the change is induced.” (183, 184)
The genotoxic potential of a substance refers to its clastogenic, aneugenic, or mutagenic potential. (p6/17 in (185) ) Clastogens are compounds that cause DNA strand breaks. Aneugens cause aneuploidy, the state of having an abnormal number of chromosomes. (186) Mutagens are substances that induce point mutations in DNA. (185)
The 2012 FDA guidance defines genotoxicity tests as “in vitro and in vivo tests designed to detect compounds that induce genetic damage by various mechanisms.” p6/35 (187) This and related documents (188) describe a standard battery of studies designed to identify genotoxic hazards consisting of:
· in vitro and in vitro assessment of chromosomal damage – including in vitro and in vivo micronucleus assays,
Both clastogens and aneugens can be detected (and distinguished) in a micronucleus test (186) which also detects spindle poisons. (189, 190) This type of test is not used to assess mutagenicity
· A test for mutagenicity, commonly the bacterial reverse mutagenicity (Ames test), (191) which has been shown to detect the majority of genotoxic carcinogens in rodents and humans.(p7/35) (187) Limitations to the test are discussed in a 1997 FDA guidance (p7/12 in (135) ) particularly where the test fails to provide “appropriate or sufficient information for the assessment of genotoxicity,” and where other types of test should also be performed. Several other mutagenicity tests have been characterized. (192)
Although well characterized, these tests can sometimes yield equivocal results with poor reproducibility or with questionable biological significance. The interpretation of these sorts of data is a complex matter, discussed in FDA guidance documents. (187, 188)
The concern that a genotoxic substance may also be a carcinogen is not without foundation. FDA’s 1996 guidance (193) states: “ Unequivocally genotoxic compounds, in the absence of other data, are presumed to be transspecies carcinogens, implying a hazard to humans.” (p5/7) The risk of carcinogenesis is typically assessed in long term rodent models or alternative methods of a shorter duration (p5/8 in (188) ) along with genotoxicity and other types of data. (188, 194)
Further discussion of how various guidelines were interpreted is found in section 12.4 .
12.2. What genotoxicity tests were performed on COMIRNATY or SPIKEVAX?
In short.: None to very few.
Dr. Marks stated : “Additionally, studies have been conducted in animals using the modified mRNA and lipid nanoparticle together that constitute the vaccine , including the minute quantities of residual DNA fragments left over after DNAse treatment during manufacturing, and demonstrate no evidence for genotoxicity from the vaccine .” (emphasis added)
This statement is contradicted by those in Pfizer’s nonclinical overview (160) (p29/36)
“No genotoxicity studies are planned for BNT162b2 as the components of the vaccine construct are lipids and RNA and are not expected to have genotoxic potential (WHO, 2005) […] Carcinogenicity studies with BNT162b2 have not been conducted as the components of the vaccine construct are lipids and RNA and are not expected to have carcinogenic or tumorigenic potential. Carcinogenicity testing is generally not considered necessary to support the development and licensure of vaccine products for infectious diseases (WHO, 2005).”
And in EMA’s EPAR assessment for Pfizer (p50/140 in (30) )
“No genotoxicity studies have been provided. This is acceptable as the components of the vaccine formulation are lipids and RNA that are not expected to have genotoxic potential.”
This statement is also contradicted by the package inserts for the two modRNA products.
· “COMIRNATY has not been evaluated for the potential to cause carcinogenicity, genotoxicity, or impairment of male fertility.” : (15, 16)
· “SPIKEVAX has not been evaluated for carcinogenic, mutagenic potential, or impairment of male fertility in animals.” (17, 195)
The question as to how the various guidelines have been interpreted is discussed in section 12.4 .
Question 184: On what basis did Pfizer “expected” that the components of the vaccine construct are lipids and RNA would not have genotoxic potential? Did FDA challenge this expectation? Q184
Dr. Marks’ footnote 3 points to the URLs for the “Summary Basis for Regulatory Action” for both COMIRNATY (47) and SPIKEVAX. (151)
Although the document cited for COMIRNATY (47) contains many redactions, the two relevant sections “Nonclinical Pharmacology/Toxicology” and “Safety and Pharmacovigilance” were not redacted. There is no reference to animal genotoxicity studies anywhere in this cited document, that support the statement “ no evidence for genotoxicity from the vaccine.” Genotoxicity studies were conducted on an excipient (see 12.3.2 ).
The document cited for SPIKEVAX similarly contains no reference to genotoxicity studies involving the “ modified mRNA and lipid nanoparticle together that constitute the vaccine.” However, s tudies with excipients or other kinds of mRNA were conducted (see 12.3.3 ).
Thus, this cited document fails to support the representation that: “studies have been conducted in animals using the modified mRNA and lipid nanoparticle together that constitute the vaccine, […] no evidence for genotoxicity from the vaccine.”
Question 185: WillFDA correct Dr. Marks’ statement that “Additionally, studies have been conducted in animals using the modified mRNA and lipid nanoparticle together that constitute the vaccine, including the minute quantities of residual DNA fragments left over after DNAse treatment during manufacturing, and demonstrate no evidence for genotoxicity from the vaccine”? Q185
12.3. What genotoxicity tests were performed on ingredients related modRNA products?
12.3.1. Relevance and reliance on supportive studies involving related modRNA products
While it is appropriate and necessary in regulatory submissions to document “supportive studies” on test articles of related composition, or involving procedurally deficient studies, sufficient detail about the studies and the compositions tested must be provided in order to weight their relevance to the current COVID-19 modRNA vaccines.
Favorable supportive data, although not independently decisive, certainly adds confidence to a favorable risk assessment. Such confidence must be tempered by the sometimes-remote comparability of the mRNA products tested to the authorized or approved product in terms of the mRNA sequence, LNP composition, method of manufacture, nucleoside modification, codon optimization, amounts and types of impurities and contaminants.
Accordingly, how these sorts of differences are considered cannot be a “one way” street. They cannot be effectively minimized when favorable supportive data bolsters confidence in an approval or authorization, but emphasized when unfavorable “supportive” impugns such confidence.
In reviewing the documents cited for COMIRNATY (47) and SPIKEVAX, (151) the reliance on nonclinical studies involving early modRNA COVID-19 vaccine prototypes, or non-COVID-19 modRNA prototypes, is evident.
Question 186: What was the regulatory basis for FDA’s heavy reliance on nonclinical studies involving early modRNA COVID-19 vaccine prototypes, or non-COVID-19 modRNA prototypes, rather than on studies involving test articles of substantially identical composition to the authorized product? To what extent was reliance based on the EUA “totality-of-evidence” standard? Q186
Question 187: How is FDA’s heavy reliance on nonclinical studies involving early modRNA COVID-19 vaccine prototypes, or non-COVID-19 modRNA prototypes, rather than on studies involving test articles of substantially identical composition to the authorized product, compatible with BLA requirements? Q187
Question 188: Which nonclinical studies have FDA requested from Pfizer of Moderna to rectify the quality and quantity of the limited studies relied upon under EUA conditions, but would have been insufficient in non-pandemic conditions? Q188
Several questions were asked earlier ( 3.6 ) regarding the studies involving COVID-19 modRNA prototypes. Supportive studies on non-COVID-19 modRNA products as well as vaccine ingredients, focusing on genotoxicity will now be considered, followed by other nonclinical studies.
12.3.2. What genotoxicity studies were performed on COMIRNATY ingredients or related modRNA products?
With a product as complex as the modRNA or adenovirus vector COVID-19 vaccines, involving DNA, RNA, lipid components spike protein, and lipid-adducts, 8.1 ) the multi-hit hypothesis of oncogenesis must be considered. (196)
It is surprising that, as described above no genotoxicity studies were conducted on COMIRNATY, or at least provided, as the EMA’s assessment report for COMIRNATY (30) states: “No genotoxicity studies have been provided. This is acceptable as the components of the vaccine formulation are lipids and RNA that are not expected to have genotoxic potential.” (p50/140)(emphasis added) This report notes a low genotoxic risk with one of the ingredients that constitute the LNP:
“The novel excipient ALC-0159 contains a potential acetamide moiety. Risk assessment performed by the Applicant indicates that the risk of genotoxicity relating to this excipient is very low based on literature data where acetamide genotoxicity is associated with high doses and chronic administration (≥1000 mg/kg/day). Since the amount of ALC-0159 excipient in the finished product is low (50 μg/dose), its clearance is high and only two administrations of the product are recommended for humans, the genotoxicity risk is expected to be very low.” (p50/140)
The absence of genotoxicity or carcinogenesis studies is reflected in the package inserts for COMIRNATY (16) “COMIRNATY has not been evaluated for the potential to cause carcinogenicity, genotoxicity, or impairment of male fertility” and SPIKEVAX. (195) “SPIKEVAX has not been evaluated for carcinogenic, mutagenic potential, or impairment of male fertility in animals.”
Question 189: While the dose of ALC-0159 appears to be low compared with the doses associated with genotoxicity (according to the EMA report), what consideration was given to a possible synergistic effect of sub-genotoxic threshold levels of this component with other vaccine components? Q189
Discussion beyond the scope of this document is required on the issue of how LNPs or their components should be defined or regulated as excipients in the context of contributing to a nanotechnology. (180, 197, 198) (see also 11.4.2 )
12.3.3. Missing genotoxicity studies of SPIKEVAX ingredients or related products ?
The number and description of the studies enumerated in this FDA document (151) ( Figure 13 ) do not match with those given in Moderna’s nonclinical overview (152) or the EMA assessment report for the Moderna product. (136) It appears that potentially unfavorable supportive data were not disclosed or adequately described in the FDA document. (151)
To compare the FDA, EMA and Moderna accounts, Table 1 of the EMA document (p43/169) has been reproduced in Table 1 . (see 10.1 ) Not included are seven pharmacology studies, of which six were identified as involving mRNA-1273, Moderna’s candidate COVID-19 vaccine.
The following Table 4 attempts to reconcile the differences between the numbers and descriptions of studies found in the two documents. The left column contains the clause-by-clause text of the paragraph under “Other Supportive Toxicology Studies” on p14/30 of the cited FDA document. (151) The right column attempts to reconcile each clause with the EMA document. More detailed discussion is given below.
Table 4 : Reconciliation of descriptions for “Other Supportive Toxicology Studies” in the FDA (151) and EMA (136) documents
|
Original text |
Comment |
|
Other Supportive Toxicology Studies The safety of SPIKEVAX is further supported by the aggregate rat repeat-dose toxicity profiles observed in six GLP toxicity studies |
Likely refers to studies identified in Table 1 as T1-T6 [see section 12.3.4 ] |
|
of five vaccines |
2 kinds of Zika, 2 kinds of CMV, hMPV/PIV3 |
|
formulated in SM-102 lipid particles containing mRNAs encoding various viral glycoprotein antigens, demonstrating tolerance of repeat doses of these vaccines without any detrimental effects. |
According to the original footnotes studies T1 and T2 employed a mixture of four lipids (“LIP4”) which were SM-102, PEG2000-DMG, cholesterol, and DSPC. The other studies T3, T4, T5, and T6 also used LIP4 in addition to “PG” which abbreviation is undefined, but possibly indicates polyethylene glycol (PEG). There are other formulation differences such as pH and buffer composition. [see section 12.3.4 ] |
|
Three other toxicology studies were also reviewed in support of safety of SPIKEVAX. |
The descriptions of the three studies alluded to in the cited FDA document (151) align only partially with the four genotoxicity studies described in the EMA report (136) (Table 1, p44/169) indicated here as studies G4. |
|
A study report from an in vitro rat micronucleus assay |
The EMA report and Moderna’s nonclinical overview (152) documents not one but two rat micronucleus studies performed on mRNA-containing test article Zika (G3) and luciferase mRNA, G4). See below. |
|
evaluating the genotoxic potential of (b) (4) mRNA in SM-102 LNP revealed no genotoxic effects of SM-102 LNP. |
The identity of the redacted mRNA is likely luciferase mRNA, with results from a Zika mRNA product not disclosed in this document. Genotoxicity signals were noted in the EMA document, albeit with qualifying language. This was not discussed in the FDA document. Moderna’s nonclinical overview described the positive result in the Zija study and the negative result in the luciferase study as “equivocal.” (p21/31) (152) See discussion below. |
|
In addition, study reports from a bacterial reverse mutation test and an in vitro mammalian cell micronucleus test of PEG2000-DMG were also reviewed. No genotoxic effects of PEG2000-DMG were observed in these studies. |
The EMA document refers to bacterial reverse mutation (G1) and in vitro mammalian cell micronucleus (G2) tests being performed on SM-102 and not PEG2000-DMG. Moderna’s nonclinical overview (p17/31) (152) documents that these two assays were performed on each LNP ingredient for a total of four tests. With study numbers 9601035 and 9601036 for the PEG2000-DMG tests. See below. The EMA document makes recommendations about mutagenic impurities in PEG2000-DMG and benzene impurities in SM-102. See below. |
The FDA document erroneously describes the rat micronucleus test as being an in vitro , not an in vivo test (compare with p50/169 of (136) ). As discussed above ( 12.1 ), this assay is not an appropriate or adequate method of assessing integration, mutagenicity, or carcinogenicity. Thus, even if it were performed on the COVID-19 mRNA vaccine, it would not address concerns about integration.
The FDA document describes only one test of this kind, on an mRNA with redacted identity. The EMA report documents two rat micronucleus studies (G3, G4 in Table 1 ) and the mRNA types as Zika (G3) and luciferase mRNA (G4). [30] This is confirmed in Moderna’s nonclinical overview obtained under FOIA. (152)
Question 190: Please confirm the identity of mystery mRNA. To the extent that redaction ever qualified for a (b)(4) exemption, the matter has already been publicly disclosed in the EMA and Moderna documents. Q190
Question 191: Please explain the reason why the genotoxicity study with the “other” mRNA was not described FDA’s document. (151) FDA’s omission does not appear to be a failure by Moderna to report the study to FDA. (152) Q191
The Moderna overview (152) (p21/31) noted that in the luciferase study (G4 in Table 1 ) the results were determined to be negative. The EMA document (136) noted that test article was “determined to be negative (non-clastogenic,)” (p50/169) further noting a transient, non-dose dependent “statistically significant decrease in poly chromatic erythrocytes” in the low dose group in male rats (p50/169 in (136) ). The EMA report also noted cytokine increases and referred to other studies showing cytokine release after IM dosing of mRNA-1273 in primates (p50/169). Moderna’s overview (152) documented that this study was not GLP compliant (p18/31), FDA failed to mention this, and the EMA were under the impression that this study was GLP-compliant. [31]
Moderna (p21/31) (152) noted positive findings in the Zika mRNA study (G3 in Table 1 ) with EMA providing more detail, noting a “statistically significant increases in micronucleated erythrocytes were reported in both sexes. A strong increase in Molecular initiating event (MIE) was observed 48 hours after the final administration in the highest dose group in male rats […]. No clear dose-response relationship was reported.” (p50/169 in (136) ).
Qualifying these findings, (p50/169) the EMA noted that non-genotoxic effects observed in toxicological studies could cause or contribute to the increase of micronucleated erythrocytes and could: affect clearance of micronucleated cells from the blood; elicit hyperthermia, increase splenic inflammation, and disturb erythropoiesis (lower reticulocyte count, higher red blood cell distribution width). Although the study details appear unavailable (see Question 209 ) the effects on erythropoiesis are unclear, but a lower reticulocyte count seen in the toxicological studies appears consistent with decreased numbers of polychromatic erythrocytes noted in the luciferase genotoxicity study. However, it is unclear from the limited detail available, how reductions in reticulocytes and polychromatic cells are mechanistically related to an increase in micronucleated cells.
Question 192: Please provide the study reports for the rat micronucleus assays conducted on the Zika (Study 9800399) and luciferase (Study AF87FU.125012 NGLPICH.BTL) test articles. Q192
Question 193: Please discuss how reductions in reticulocytes and polychromatic cells seen in toxicology and genotoxicity studies are mechanistically related to an increase in micronucleated cells seen in the Zika mRNA genotoxicity study. Q193
Question 194: Do reductions in reticulocytes and polychromatic cells, as well as disturbances in erythropoiesis raise any concerns for bone marrow toxicity? What follow up studies or risk analysis has FDA requested on this topic? Q194
The EMA, in considering these studies as well as other factors such as dosing concluded that “Taking all these data together, a relevant genotoxic risk is thus not expected for mRNA-1273.” (p 55/169) (136)
Moderna’s approach was similar, although providing no comment on the effects seen on reticulocytes, polychromatic cells and erythropoiesis :
“The equivocal results are likely driven by micronuclei formation secondary to increased cytokines and/or body temperature induced by LNP-driven systemic inflammation at high systemic (intravenous) doses. Overall, the genotoxic risk to humans is considered to be low due to minimal systemic exposure following IM administration, limited duration of exposure, and negative in vitro results.” “(p21/31) (152)
Because assumptions that distribution is almost exclusively LNP dependent and largely independent of mRNA sequence are at best tenuous ( 11.4.2 ), reliance on favorable toxicological and genotoxicity findings is not firmly grounded.
In sum, the studies described fail to “demonstrate no evidence for genotoxicity from the vaccine,” as represented. Rather, genotoxicity signals were observed but not disclosed in the FDA document (151) or in the Marks response. Although the studies were not performed on “…the modified mRNA and lipid nanoparticle together that constitute the vaccine,“ as represented, the studies need to be fully reported regardless of any qualifying language. Even if the qualifying language is justified, the signal still contributes to the body of evidence related to concern for genotoxicity.
Question 195: Given the disclosure (11.4.1) that the sizes of LNPs in a CMV vaccine examined in a biodistribution study were smaller than those found in mRNA-1273, despite misleading statements suggesting that they were the same, what assurances can FDA give that the formulations of the Zika and luciferase mRNA vaccines subjected to genotoxicity tests were identical to mRNA-1273, other than in the modRNA sequence coding for the target antigen/ luciferase? Q195
The FDA report notes the conduct of bacterial reverse mutation and in vitro mammalian cell micronucleus test on the PEG2000-DMG LNP component. However, the EMA document ( Table 1 , studies G1, G2) refers to these tests being conducted on the SM-102 component and not PEG2000-DMG. Moderna’s nonclinical overview (p17/31) (152) documents that these two assays were performed on each LNP ingredient, [32] for a total of four genotoxicity tests.
Question 196: Will FDA resolve the discrepancies between the FDA, EMA and Moderna documents regarding which LNP components were tested in these genotoxicity tests. Q196
Although these tests were conducted on LNP components and not Drug Product, subthreshold genotoxic potential may be additive or synergistic with any effects from other vaccine components or impurities. It is therefore of note that the EMA recommends (p25/169) that “Potential presence of mutagenic impurities in PEG2000-DMG should be evaluated and the results will be provided post-approval.”
Question 197: What was the nature of the concern for mutagenic impurities in PEG2000-DMG? Q197
Question 198: Did FDA have a similar concern for mutagenic impurities in PEG2000-DMG as did EMA? Q198
Question 199: How was this concern for mutagenic impurities in PEG2000-DMG resolved? Q199
Question 200: Given that the evaluation of the potential presence of mutagenic impurities in PEG2000-DMG was to be provided post-approval, when exactly did this occur? Q200
Question 201: If mutagenic impurities did exist in PEG2000-DMG prior to resolution of this issue, how many doses of mRNA-1273 (and to how many people) had been administered either in clinical trials or post approval/ authorization? Q201
Question 202: Has any risk assessment been conducted or requested by FDA to assess whether synergistic effects occurred between any subthreshold mutagenicity of impurities in PEG2000-DMG and any effects from other vaccine components or impurities? Q202
Two similarly relevant EMA recommendations were made regarding SM-102 (p165/169).
“c) A risk assessment for the presence of benzene in SM-102 should be completed and the control strategy should be updated and submitted no later than 30-06-2021.
g) Mutagenic impurities, including impurities originating from the starting materials, have not been discussed, discussion with regard to ICH M7 should be provided. Special attention should be set on the potential mutagenic primary halogens (i.e. starting material SM-102-11). “
Question 203: What was the nature of the concern for benzene or mutagenic impurities in SM-102? Q203
Question 204: Did FDA have a similar concern for benzene or mutagenic impurities in SM-102 as did EMA? Q204
Question 205: How was this concern for benzene or mutagenic impurities in SM-102 resolved? Q205
Question 206: Given that the risk assessment for the presence of benzene in SM-102 was to be provided by June 30 2021, when exactly did this occur? Q206
Question 207: If benzene or mutagenic impurities did exist in SM-102 prior to resolution of this issue, how many doses (and to how many people) of mRNA-1273 had been administered either in clinical trials or post approval/ authorization? Q207
Question 208: Has any risk assessment been conducted or requested by FDA to assess whether synergistic effects occurred between any benzene and subthreshold mutagenicity of impurities in SM-102 and any effects from other vaccine components or impurities? Q208
12.3.4. Questions regarding nonclinical studies other than genotoxicity
In reviewing the documents cite by Dr. Marks for COMIRNATY (47) and SPIKEVAX, (151) a number of questions arise regarding procedurally deficient or other supportive nonclinical studies. See also questions in section 3.6 regarding COVID-19 modRNA prototypes and section 12.3.1 on the relevance and reliance on supportive studies involving related modRNA products.
The six repeat dose toxicity studies on five Moderna vaccines referred to in the FDA document (151) as “supportive” correspond to studies T1-T6 identified in Table 1 . The EMA (136) identifies two kinds of Zika, two kinds of CMV, and one hMPV/PIV3 mRNA product. The comparability with authorized product of method of manufacture, non-coding regions, codon optimization, use of N1-methyl pseudouridine and type and amount of impurities, is unknown. There are other formulation differences such as pH and buffer composition. In some of the studies the ingredient “PG” was used, but this abbreviation is not defined in the EMA document. (136)
Question 209: Will FDA release the full original reports for all Moderna and Pfizer toxicological studies supporting the various EUA or BLA’s for their COVID-19 mRNA vaccines? Q209
Question 210: What is the identity of the ingredient “PG” used in some of Moderna’s repeat-dose toxicology studies and in its biodistribution study? Is this polyethylene glycol (PEG)? Q210
Question 211: Given the disclosure (11.4.1) that the sizes of LNPs in a CMV vaccine examined in a biodistribution study were smaller than those found in mRNA-1273, despite misleading statements suggesting that they were the same, what assurances can FDA give that the formulations of the Zika and luciferase mRNA vaccines subjected to genotoxicity tests were identical to mRNA-1273, other than in the modRNA sequence coding for the target antigen/ luciferase? Q211
None of the above studies involve an mRNA encoding for SARS-CoV-2 spike protein. However, one non-GLP repeat-dose immunogenicity and toxicity study of mRNA-1273 in rats is described in the FDA document under the general heading “Nonclinical Pharmacology/Toxicology.” The FDA document notes that: “Findings from the intramuscular repeat dose rat toxicity study demonstrated that vaccine doses of up to 100 mcg (two doses given three weeks apart) were well-tolerated.”
This study appears to correspond with study T7 ( Table 1 ) in the EMA document. However, absent from the FDA document is mention of “major procedural/ methodological limitations” (p48/169 in (136) )) indicated in the EMA document.
“The product-specific Study 2308-123 was not conducted in GLP-compliance, and exhibits major procedural/methodological limitations. In principle these aspects would render this study inadequate for evaluating the repeated dose toxicity of mRNA-1273 to the extent recommended in relevant guidance on non-clinical development of vaccine products.” (p48/169 in (136) )
Question 212: Was FDA aware of the procedural/methodological limitations in Moderna’s only repeat-dose toxicology study on its SARS-CovV-2 mRNA candidate described in the EMA document? Q212
Question 213: The procedural/methodological limitations indicated by EMA for Moderna’s only repeat-dose toxicology study on its SARS-CovV-2 mRNA candidate appear to extend beyond non-compliance with GLP. What was the nature of these limitations? Q213
Question 214: The absence in FDA’s document of a qualifying statement similar to that in the EMA document appears to be a material omission possibly affecting the interpretation of the body of nonclinical data. Please justify or comment. Q214
Despite the qualifying language that “ render this study inadequate,” EMA concluded: “ However, as no clear differences in toxicity are observed between study 2308-123 [i.e. on mRNA-1273] and the repeated dose toxicity studies conducted with other LNP-mRNA products, the latter studies are considered sufficient to support clinical development and MAA” (p48/169)
Question 215: What standard of evidence did FDA ascribe to the comparison of data from a procedurally, methodologically, and inadequate study of Moderna’s SARS-Cov-2 vaccine candidate to data from studies on other mRNA products? Q215
Question 216: Even if, per Question 215, the quality of data from the only repeat-dose toxicology study of Moderna’s COVID-19 vaccine candidate was sufficient to meet the EUA “totality of evidence” standard, did FDA consider that this study met BLA requirements? Q216
Some of Pfizer’s other nonclinical studies are described in EMA’s 2021 report (30) including a repeat dose toxicity study in rats (P49/140). “Large unstained cells,” presumably in blood, were observed, but EMA’s report noted that: “The applicant also informed that characterisation of large unstained cells was not conducted since the identification of these cells does not provide additional information. The CHMP found this agreeable.”
The report elaborates on p50/140:
“Haematology: At 30ug BNT162b2 V9 and 100ug BNT162b2 V8, there was a moderate to strong reduction of reticulocytes (48-74%, not specified for V9) coupled to lowered red cell mass parameters (RBC, HGB, and HCT). There was a moderate to strong increase (>100%) in large unclassified cells [LUC], neutrophils, eosinophils, basophils and fibrinogen that may be related to the inflammatory/immune response. The changes were reversible. No effects on coagulation were observed for V8 whereas a slight increase in fibrinogen was observed with V8 and V9.”
Question 217: Were these large unstained cells in Pfizer’s repeat dose toxicity study reported to FDA?Did FDA seek clarification as to their nature? What was Pfizer’s response? Q217
Question 218: Referencing Question 61 (section 3.6), what other differences in the results from nonclinical studies were found between the V8 and V9 Pfizer product versions? Q218
Further examination of Pfizer’s nonclinical data disclosed under FOIA, for example, (199) is warranted.
12.4. Generous interpretation of WHO guideline vaccine exemption for genotoxicity or carcinogenicity studies
A number of guidelines have been issued that speak to the sorts of genotoxicity or carcinogenicity studies that might be required for these products. Ambiguous, or sometimes apparently conflicting statements, may have provided justification for the relative dearth of studies that were conducted.
Although not applying to biologics. The FDA guidance on genotoxicity testing (187) provides instructive general principles: “Nevertheless, a battery approach is still reasonable because no single test is capable of detecting all genotoxic mechanisms relevant in tumorigenesis.” (p 6/35 ) The same can be said for a guidance on the mutagenicity of impurities (184) “Specifically, changes should be evaluated to determine if the changes result in any new mutagenic impurities or higher acceptance criteria for existing mutagenic impurities .” (p9/38)
The 2007 WHO guideline on DNA vaccines (49) discusses the need for case-by-case consideration in the design of the nonclinical safety program, which should be performed on every novel vaccine:
“In designing the nonclinical safety programme for a DNA vaccine product, the 2007 WHO guidelines on nonclinical evaluation of vaccines should be consulted in addition to the guidance provided here . […] Every product should be evaluated on a case-by-case basis . As a general rule , nonclinical safety assessment should be performed on every novel vaccine or vaccine/adjuvant formulation.” (p20/25 in (49) ) (emphasis added)
This same document (49) continues (p23/25) with an apparently mixed message: “The standard battery of genotoxicity and conventional carcinogenicity studies is not applicable to DNA vaccines. However, genotoxicity studies may be required to address a concern about a specific impurity or novel chemical component, e.g. a complexing material that has not been tested previously.”
Introducing a discussion of nonclinical evaluation of mRNA vaccines and citing the WHO 2005 (177) and 2014 (172) documents, the 2021 WHO guideline [33] (71) lists a number of issues for which agreement should be reached with regulators on the types of nonclinical studies to be conducted, rather than reliance on a general exemptive rule.
“ Because of the novelty of this product class and for the sake of inclusiveness, numerous issues are listed in this section. Not all of these issues will necessarily be relevant to mRNA vaccines, depending on their design. However, it is incumbent upon the vaccine developer/manufacturer to provide evidence demonstrating the proof-of-concept (for example, immunogenicity and challenge protection) and safety of their candidate vaccine. The types, design and number of studies expected should be agreed with the NRA .” (p45/66) Among the issues listed where this sort of agreement should be reached are “Biodistribution and persistence” studies. (p46/66 in (71) )
There is no discussion of the lenient interpretation of WHO guidelines concerning the conduct of genotoxicity or carcinogenicity studies for vaccines. Genotoxicity is mentioned with regard to LNPs, lipid and other novel excipients.
“d. Novel lipids and novel LNPs: because the lipids used to formulate the LNPs affect the overall charge of the particle, when using LNPs made with novel lipids or when the LNPs are themselves modified (for example, altered ratios or modified processes) and these LNPs have not previously been nonclinically and clinically tested in mRNA products encapsulated in LNPs then evaluation of the toxicity of the new formulation containing the novel lipids (or any novel excipients) may be required. Furthermore, the NRA may require that the genotoxicity and systemic toxicity of the novel lipid component be assessed, similar to the expectations for novel adjuvants set out in the WHO Guidelines on the nonclinical evaluation of vaccine adjuvants and adjuvanted vaccines (16) and/or those for new chemical entities in the ICH guideline S2 (R1)” (58) .
“e. Novel formulations : likewise, for formulations (other than LNPs) containing novel excipients, data on and assessment of the systemic toxicity and genotoxicity of the formulation may be expected.”
A 2014 WHO guidance on adjuvants p27/42 (172) delivers a different message: “Genotoxicity studies are normally not needed for the final vaccine formulation (1). However, a standard battery of genotoxicity studies is generally recommended for most novel adjuvants that are (or contain) new chemical entities (31, 37). Based on previous experience, carcinogenicity studies are generally not needed for adjuvants or adjuvanted vaccines.”
An FDA 2010 guidance on cell substrates for vaccine production (63) (p38/50) states:
“If the presence of an oncogenic virus is suspected because of the cell phenotype or the origin of your cell substrate, we recommend that you perform oncogenicity testing in animals using lysates of the cell substrate. For cell substrates with a tumorigenic phenotype, we recommend that you perform oncogenicity testing in animals using DNA from the cell substrate in order to provide assurance that residual DNA is non-oncogenic (also see Section III.C.6). Oncogenicity testing might also be appropriate for some products with high quantities of residual cellular DNA .” (emphasis added)
The FDA 1996 guidance (193) on the factors to consider for carcinogenicity studies of drugs has been discussed above, ( 12.1 ) particularly its statement (p 5/7) “ Unequivocally genotoxic compounds, in the absence of other data, are presumed to be transspecies carcinogens, implying a hazard to humans.”
The 2005 WHO guidance (177) on nonclinical evaluation of vaccines relied upon by Pfizer (section 12.2 ) states:
“ Genotoxicity studies are normally not needed for the final vaccine formulation. However, they may be required for particular vaccine components such as novel adjuvants and additives . If needed, the in vitro tests for mutations and chromosomal damage should be done prior to first human exposure. The full battery of tests for genotoxicity may be performed in parallel with clinical trials [citing (200) [34] , appears same as (135) ] Carcinogenicity studies are not required for vaccine antigens . However, they may be required for particular vaccine components such as novel adjuvants and additives.” (p20/36 in (177) )(emphasis added)
Similar to our discussion of the need for kinetics studies ( 11.2.3 ), this2005 WHO guideline speaks of classical vaccine antigens, not gene therapy based produced. It also states that these studies are not “normally” needed, but may be required for “ novel adjuvants and additives.”
Taken on its own, it appears this has been generously interpreted by Pfizer. Taking the totality of other guidelines described, even with some of the ambiguities, there appears to be little justification for the assertion that these studies were not needed.
Question 219: Did FDA challenge Pfizer’s assertion based on WHO 2005 guideline, that genotoxicity would not be needed? Q219
Question 220: Will FDA work to remove ambiguities in their own guidelines related to the conduct of genotoxicity and carcinogenicity studies for modRNA pro-vaccines? Q220
13. Cancer signals
Dr. Marks stated: “Pharmacovigilance data in hundreds of millions of individuals also indicate no evidence indicative of genotoxicity […] we now have access to global surveillance data on over one billion doses of the mRNA vaccines that have been given, and there is nothing to indicate harm to the genome, such as increased rates of cancers.”
He also testified before the House Select Subcommittee on the Coronavirus Pandemic on February 15, 2024, that “we have not detected any increase in cancers with the Covid-19 vaccines.” (1 hr 55 mins in (201) ) This statement is inconsistent with the 23 Proportional Reporting Ratio signals found by CDC’s Disproportionality Signal Analysis disclosed under FOIA (202, 203) ( Figure 18 ) for some cancer types or codes. Adjusting for masking and filtering, this could extend to 59 signals. (204) Although these statistical signals have not yet been shown to be causal, they represent potential risk the FDA was required to consider for the granting of an EUA.(Sec 564 ©(2)(B) in (205) )
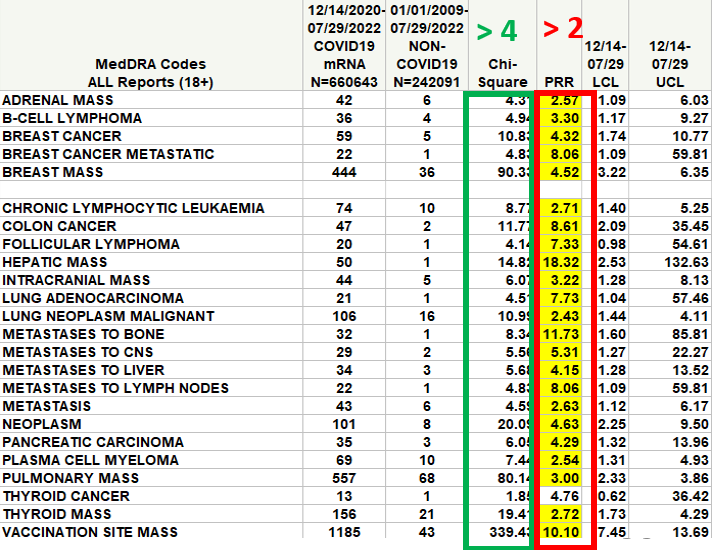
Figure 18 : Cancer Signals from CDC’s July 2022 PRR analysis
Using UK statistics Alegria and Nunes (206) found a large increase in morbidity (disabilities) and mortality due to malignant neoplasms starting in 2021 and accelerating in 2022, for individuals aged 15 to 44 in the UK. Similar trends were found in US Statistics. (207) Before a Texas Senate committee, Wiseman discussed a number of potential mechanisms for this COVID-19 era related increase (208) related to the effects of social distancing and the SARS-Cov-2 virus, particularly the spike protein. An effect of the COVID-19 pro-vaccines cannot be excluded.
Question 221: Although FDA did not identify any cancer signals using Empirical Bayesian Data Mining, (EBDM) their analysis was recently suggested to be seriously flawed. (204) Please provide full details of all signals generated by EBDM, including where the threshold is set to EB05>1, and the RGPS procedure within the Empirica software is used to adjust for masking, using the ER05>1 criteria? Q221
14. modRNA vaccines elicit cryptic uncharacterized frameshift proteins of unknown toxicity
Although not a subject of Dr. Ladapo’s concerns, this related matter warrants attention.
A paper was published by Mulroney et al . (209) in Nature entitled: “ N1-methylpseudouridylation of mRNA causes +1 ribosomal frameshifting.” The paper was accompanied by a press release (210) from the University of Cambridge whose scientists collaborated with others from the universities of Oxford, Liverpool and Dublin, and elsewhere. A number of these labs act as centers within the UK’s Medical Research Council (MRC) or National Institute for Health and Care Research (NIHR).
The authors synthesized a pool of peptides whose sequences were predicted by +1 shifts of the reading frame for spike protein in vaccinal BNT162b2 modRNA. Murine spleen and human peripheral blood mononuclear cells were taken after vaccination with the Pfizer or Astra-Zeneca products and exposed to these frameshift peptides in an in vitro assay of T cell function. Cells from mice or humans previously vaccinated with the Pfizer but not the Astra-Zeneca product responded in this assay. According to the press release about a third of the people responded in this way, indicating that they had “seen” antigens resembling those peptides before.
The paper also discussed how frameshifting would likely elicit a family of chimeric proteins consisting of portions of in-frame and out of frame amino acid sequences. The authors showed that frameshifting was likely to occur at sites rich in N1-methylpseudouridine, engineered into the RNA to produce the “nucleoside modified” RNA (modRNA) to evade innate immune attack. Since the viral-vector DNA Astra-Zeneca vaccine did not contain this modification, no evidence of exposure to frameshift proteins was detected in the cells of mice or people given this product. Frameshifting in a model system occurred in approximately 10% of ribosomal reads which could be reduced by limiting the extent of N1-methylpseudouridylation at particular loci.
The authors concluded that “these data suggest that vaccination with 1-methyl. mRNA can elicit cellular immunity to peptide antigens produced by +1 ribosomal frameshifting in both major histocompatibility complex (MHC)-diverse people and MHC-uniform mice.”
One of the authors was quoted (210) as saying: (emphasis added)
“We can remove the error-prone code from the mRNA in vaccines so the body will make the proteins we want for an immune response without inadvertently making other proteins as well. The safety concern for future mRNA medicines is that mis-directed immunity has huge potential to be harmful , so off-target immune responses should always be avoided.”
With this and similar statements, the authors state in the paper that “Although there is no evidence that frameshifted products in humans generated from BNT162b2 vaccination are associated with adverse outcomes. ” This statement appears to be based on the observation that none of the 21 subjects providing samples had “ reported undue effects as a result of vaccination.” It is unclear how it is possible to draw any conclusions about the safety implications of the frameshifted proteins given that:
· Only a small number of vaccinated subjects (n=21) provided samples for this study.
· The use of subjects who had not reported adverse outcomes constitutes a selection bias.
· This was not a controlled trial.
The stated premise for the study was: “ So far, no study has investigated the fundamental question of whether modified ribonucleotides can affect the maintenance of the correct reading frame during translation of a synthetic transcript.” If these authors were able to predict the existence of frameshifted proteins, why were Pfizer’s scientists unable to do so? Certainly, the possibility of “cryptic proteins” being translated from alternative out of frame open reading frames was known to the co-founder of Moderna in a 2016 paper. (p326 of (67) )
The same question may be asked of regulators, especially in light of unresolved concerns raised by EMA (p137/140 in (30) ) such as “when present in the cell it cannot be excluded that different proteins than the intact full-length spike will be expressed” as well as discrepancies reflected in EMA’s Specific Obligation regarding the identities of Western Blot (WB) bands obtained by in vitro expression assays. (30) (see 3.3 above). Could some of these bands represent chimeric proteins containing both frameshift regions and in-frame regions still capable of reacting with the anti-spike antibodies used in the Western blots? Could these bands represent other kinds of cryptic proteins (sometimes called cryptides or crypteins) such as those produced from alternate start sites (67, 211) or proteolytic cleavage. (212)
According to 2021 WHO guidelines for mRNA vaccines, (71) manufacturers should provide details of “ unexpected ORFs” (Open Reading Frames): “The complete annotated sequence identifying all ORFs (including any unexpected ORFs) and all other sequence elements (including their justification for use) should be provided.” (p13/66) (emphasis added) Certainly, frameshifted sequences would qualify as “ unexpected ORFs ”
Although this study involved the Pfizer vaccine, due to the involvement of the same nucleoside modification deployed in the Moderna product, frameshifting is also likely for that product.
Furthermore, the covalent lipid–RNA adducts and truncated RNA contained within the LNPs reduce the effective mRNA dose available for translation. These modifications can also perturb ribosome progression, potentially also contributing to ribosomal frameshifting (~8% observed in Mulroney et al.), which may generate incomplete or altered proteins. At the same time, residual LNP components and misprocessed RNA fragments can potentially increase cellular stress and immunogenicity. (213) Together, these effects effectively narrow the therapeutic window of mRNA-LNP vaccines, creating a disconnect between nominal “dose” and functional antigen delivery.
Question 222: Did Pfizer or Moderna identify within their modRNA sequences any unexpected ORFs, including frameshift sequences per the WHO guidelines (71)? What were these sequences and when was this information provided? Q222
Question 223: Given the submission and publication of the Mulroney paper in January and December 2023 respectively, when did FDA first learn about the findings in this paper and from whom? Q223
Annex 3 (p115/178) of a 2022 WHO Expert Committee dealing with the quality, safety and efficacy of mRNA vaccines (214) considered the formation of “neo-antigens or unwanted immune responses:” (emphasis added):
“Demonstration of expression of the complete encoded protein(s) without truncated or alternative forms should be provided. In particular, if expression of truncated or alternative forms of the target antigen is demonstrated during characterization studies and these alternative forms would result in neo-antigens or unwanted immune responses , then this may require a redesign of the mRNA sequence. “
The 2007 WHO guideline, (49) albeit addressing DNA vaccines notes that the “long-term expression of a foreign antigen may result in an undesired immunopathological reaction.”
Question 224: Given the production of neo-antigens or unwanted immune responses that “may require a redesign of the mRNA sequence” according to this WHO committee (214) and the description of the modRNA vaccines as “error-prone” in the Mulroney press release, (210) does FDA consider the non-selective N1-methylpseudouridylation of the Moderna and Pfizer COVID-19 vaccines to be an inherent design flaw? Q224
Question 225: What discussions have taken place between Pfizer or Moderna and FDA or other government agencies or entities regarding the need to redesign the COVID-19 vaccines as well as other vaccines employing the same technology? Q225
Question 226: What will be the regulatory pathway for the introduction of redesigned modRNA-based vaccines? Q226
Question 227: Is FDA attempting to characterize the frameshift proteins in terms of their primary, secondary and tertiary structures, their glycosylation patterns, the sites and kinetics of their production, their pharmaco- and toxico-kinetics? Q227
Question 228: What efforts have been made or are are underway to study the identity, pharmacology and toxicology of these frameshift proteins? Q228
Question 229: What studies have been conducted and which in silico tools been utilized to screen for likely interactions of these frameshift proteins in the body? Q229
Question 230: In addition to in vitro and animal models, are these studies being conducted in humans? How do the actions of the frameshift proteins vary by age, gender, genetic make-up and comorbidity? Q230
Question 231: Given their possible chimeric nature, what efforts are underway to determine whether there are synergistic pharmacologic, immunologic or toxicologic effects between the frameshift proteins and the intended in-frame spike proteins? Q231
Question 232: Have genomic and proteomic databases and tools such as BLAST been interrogated to determine if there are any homologies between the proposed frameshift proteins and peptides and known proteins? Q232
Question 233: What efforts are underway to determine if there are associations between the formation and type of frameshift proteins and adverse events that have been already been experienced or reported? Q233
Question 234: What efforts are underway to determine if there are likely to be long-term consequences of these frameshift proteins? Q234
Question 235: What efforts are underway to monitor for the occurrence of long-term consequences of these frameshift proteins? Q235
Question 236: What efforts are underway to determine methods for the diagnosis of possible harms caused by frameshift-proteins and treatments to limit, prevent or treat harm? Q236
The leaked EMA 2020 document cited in 3.4 (p71/78) raises a question “ regarding the risk of autoimmune responses induced by the modRNA” and invites the applicant “to further discuss the possibility that the mRNA vaccine can trigger potential autoimmune responses and how do it plan [sic] to possibly evaluate their occurrence. ([confidential information deleted])””
Question 237: Did FDA express the same concern as EMA regarding vaccine-elicited autoimmune responses? Did Pfizer or Moderna submit a discussion on this topic to FDA, per the invitation extended to Pfizer in the EMA document? When? Please supply a copy. Q237
FDA’s 2007 guidance on plasmid vaccines (64) , although downplaying the likelihood of autoimmune reactions (see 4.6 ) noted “ Yet the possibility persists that DNA vaccines might idiosyncratically cause or worsen organ-specific autoimmunity by encoding antigens (including cryptic antigens) that cross-react with self.” (p8/13)
The document proposes (p8/13) a course of action in the event that “ an immune response is induced by a transgene product encoding self-antigen.” (emphasis added)
“In cases where an immune response is induced by a transgene product encoding self-antigen (such as a cytokine, chemokine, surface receptor/ligand, or cryptic self-antigen), we recommend that you examine potential cross-reactivity with the corresponding endogenous protein. If a persistent immune response against an endogenous protein is detected, we recommend that you evaluate potential adverse effects by studying the analogous animal gene in a relevant animal model. We further recommend that you monitor whether an immune response against the self-antigen is elicited during the clinical trial, and carefully evaluate the effect of this response on trial participants.”
In this case where immune reactions have been detected to model frameshift proteins, a similar course of action is appropriate.
According to the Mulroney paper or its associated press release, the immune responses elicited by the “ unintended proteins ” could have a “ huge potential to be harmful” with “ unintended side-effects.” These responses are described as “ unintended,” “off-target,” or “mis-directed” immune responses. Given the concern expressed by EMA, as well as the unknown homologies ( Question 232 ) between the frameshift proteins and known proteins, such immune responses could be autoimmune in nature.
Question 238: In view of the evidence for off-target immune responses elicited by frameshift proteins described by Mulroney et al., as well as the concerns expressed in WHO and EMA documents, have Pfizer or Moderna been asked to submit, or have Pfizer or Moderna already submitted any risk assessments related to the production of frameshift proteins? When? Please supply a copy. Q238
Question 239: Is FDA aware of, or has it solicited or received from Pfizer, Moderna or other research entity, a full characterization of the off-target immune response elicited by frameshift proteins, alone or in combination with on-target proteins? In addition to characterization of the cellular response, as was partly provided in the Mulroney paper, does this work also include a characterization of the humoral response, which was not described by Mulroney et al. Q239
The package insert at the time of the Mulroney paper for COMIRNATY stated (15) : “Each 0.3 mL dose of COMIRNATY (2023-2024 Formula) is formulated to contain 30 mcg of a nucleoside modified messenger RNA (modRNA ) encoding the viral spike (S) glycoprotein of SARS-CoV-2 Omicron variant lineage XBB.1.5 (Omicron XBB.1.5).” (emphasis added)
The package insert for SPIKEVAX stated: (17) “ Each 0.5 mL dose of SPIKEVAX (2023-2024 Formula) contains 50 mcg nucleoside-modified messenger RNA (mRNA) encoding the pre-fusion stabilized Spike glycoprotein (S) of the SARS-CoV-2 Omicron variant lineage XBB.1.5.” (emphasis added)
Question 240: What actions will FDA be taking to correct these potentially misleading statements by including appropriate labelling language describing the production of uncharacterized off-target frameshift proteins with unknown toxicology capable of eliciting uncharacterized off-target immune response of yet unknown clinical significance? Q240
Question 241: What other steps will FDA take to inform the medical community and the lay public of he potential risks associated with the production of frameshift proteins? Q241
Question 242: What analysis has FDA conducted, or will conduct to investigate the root causes and systems failures for their apparent failure and/or that of the manufacturers to identify, detect, report, and investigate the formation of frameshift proteins and their potential risks? Q242
Question 243: If appropriate, what corrective actions has or will FDA implement within its own organization to ensure that this failure will not be repeated? Q243
Question 244: If appropriate, what regulatory actions has or will FDA implement regarding the manufacturers of modRNA COVID-19 vaccines to ensure that this failure will not be repeated? Q244
FDA has previously failed to insist on the study and assessment of risk of the pharmacology and toxicology of novel spike protein heterotrimers forming after injection of the bivalent COVID-19 modRNA vaccines. (142) The evidence for the elicitation by modRNA COVID-19 vaccines of uncharacterized frameshift proteins represents a developmental and regulatory failure to ask fundamental questions that could affect the safety and effectiveness of, and confidence in, these products. This failure can hardly be consistent with the claims that the review of these products has met “rigorous scientific standards,”
As Dr. Marks noted, over a billion doses of the modRNA vaccines have been administered. The potential is therefore very great for past or future harm due to the heretofore undetected elicitation of uncharacterized off-target frameshift proteins with unknown toxicology capable of eliciting uncharacterized off-target immune response of yet unknown clinical significance.
Question 245: Have Pfizer or Moderna submitted any data or risk analysis concerning the possible formation of other kinds of cryptic proteins (cryptides or crypteins) such as those produced from alternate start sites(67, 211) or proteolytic cleavage.(212) Q245
Question 246: Along the lines of the questions enumerated above, will FDA conduct full assessments of past or future harms associated with these proteins, identity root causes for the failure to identify this problem sooner, identify corrective actions to prevent future failures, and to inform the public of these findings? Q246
Question 247: Will FDA conduct or support the development of methods for the diagnosis, prevention and treatment of harm related to frameshift or other possible cryptic proteins? Q247
15. COVID-19 pro-vaccines meet FDA definition of gene therapy
(Adapted from (215) ) The COVID-19 pro-vaccines meet FDA’s definition of g ene therapy products. (216)
(emphasis added) “Human gene therapy/gene transfer is the administration of nucleic acids , viruses, or genetically engineered microorganisms that mediate their effect by transcription and/or translation of the transferred genetic material, and/or by integrating into the host genome. Cells may be modified in these ways ex vivo for subsequent administration to the recipient, or altered in vivo by gene therapy products administered directly to the recipient .”
A similar expanded definition is given in FDA’s Guidance on Long Term Follow-Up After Administration of Human Gene Therapy Products. (217) Both this and an earlier guidance (218) for the “Preclinical Assessment of Investigational Cellular and Gene Therapy Products” states:
“This guidance does not apply to therapeutic vaccines for infectious disease indications that are typically reviewed in CBER/Office of Vaccines Research and Review (OVRR)”
Moderna, Inc., the maker of a mRNA Covid-19 vaccine, acknowledged in their 2Q 2020 SEC filing (219) [35] thus ”Currently, mRNA is considered a gene therapy product by the FDA.” Further, the founder of BioNTech in a 2014 paper (66) stated “ One would expect the classification of an mRNA drug to be a biologic, a gene therapy or a somatic cell therapy. “
Since these agents are Gene Therapy products, long term surveillance is warranted for delayed malignant, neurologic, autoimmune, hematologic, other disorders or effects on the genome or gene expression. This is reflected in FDA’s guidance document “ Long Term Follow-up After Administration of Human Gene Therapy (GT) products.” (217) The length of monitoring advised by FDA may be (emphasis added) “ as long as 15 years following exposure to the investigational GT product, specifying that the LTFU observation should include a minimum of five years of annual examinations , followed by ten years of annual queries of study subjects, either in person or by questionnaire.”
Accordingly, the designation of these vaccines as Gene Therapy products is not merely a semantic nicety; rather it has regulatory consequences in terms of the long term follow up manufacturers should be required to conduct.
Question 248: Will FDA acknowledge the biological reality of these products and regulate them as gene therapies? Q248
16. Conclusion
Dr. Marks stated: “We would like to make clear that based on a thorough assessment of the entire manufacturing process, FDA is confident in the quality, safety, and effectiveness of the COVID-19 vaccines. The agency’s benefit-risk assessment and ongoing safety surveillance demonstrate that the benefits of their use outweigh their risks.”
It is difficult to share this confidence, given the analysis in this document, as well as our (DMW) recent analysis (204) of safety signal data that found evidence that hundreds of safety signals were missing from FDA’s EBDM analysis of VAERS data.
Our questions, although not intended to be exhaustive, reflect a concern that the available data are inconsistent with the central message of the public health COVID-19 vaccination campaign, which portrays the COVID-19 vaccines as “meeting rigorous scientific standards” and as being established as “safe and effective.” These terms appear ubiquitously, for example, on CDC’s web site (220) , representing that the “vaccines are safe and effective” and that they “met the Food and Drug Administration’s (FDA’s) rigorous scientific standards for safety, effectiveness, and manufacturing quality needed to support emergency use authorization (EUA). ”
As per statute and regulation, Emergency Use Authorization (EUA) (34) does not require a new drug to be established as “safe and effective” as in a conventional approval. Rather, an EUA requires a lower standard whereby “ based on the totality of the scientific evidence available, it is reasonable to believe that the product may be effective.” The “totality” standard allows the FDA to consider evidence of a lower type or procedural or statistical quality not normally considered in a conventional approval.
The safety standard for an EUA requires that “the known and potential benefits of the product, […] outweigh [its] known and potential risks. “ Uncertainties, irregularities, or deviations from normal accepted practice in the manufacturing process represent “potential risks” FDA was required to consider. It was not necessary to prove that DNA contamination, the SV40 sequence, or frameshifted proteins did caused harm, the mere fact of these phenomena constituted an uninvestigated potential risk FDA was legally required to consider when granting the EUAs.
Given the rapid launch of the COVID-19 pro-vaccines, manufacturing and safety testing standards needed to be tightened, not loosened as described here, adding potential, if not actual risk.
Generous interpretation of various guidelines, and the word “expects” has been used by both sponsors and regulators to justify why a certain test should not be done or a safeguard implemented, without providing detailed justification supporting the assertion, as in these examples: (bold added)
· “mRNA vaccine is expected to remain in the cytoplasm” ( 4.3.1 ) (71)
· “residual DNA is expected to degrade rapidly and has a very low likelihood of reaching the nucleus” (126)
· “FDA expects that much of the manufacturing process and controls, as well as the facilities for vaccine production, for the modified COVID-19 vaccine will be identical to that of the prototype COVID-19 vaccine.” . (149)
· “The protein encoded by the RNA in BNT162b2 is expected to be proteolytically degraded like other endogenous proteins .” (160)
· “Adjuvants are expected to exert their action locally in close connection to the antigen.” (172)
· “No genotoxicity studies are planned for BNT162b2 as the components of the vaccine construct are lipids and RNA and are not expected to have genotoxic potential (WHO, 2005) “ (160)
The developmental and regulatory failure that the findings related to residual DNA and frameshift proteins represent are hardly evidence that these modRNA vaccines meet any “ rigorous scientific standards,” and will no doubt erode further the trust in public health institutions. (221)
There are many more questions to ask. One critical area to consider, as described in a policy brief by Gutschi and Seger, (222) is the view that LNPs as passive excipients do not reflect their demonstrated biological activity, adjuvant-like properties, or unpredictable biodistribution. Regulatory frameworks must evolve to treat LNPs as active, bioactive, transfection agents, subject to the same level of scrutiny as other pharmacologically active agents.
We hope that the questions posed here will allow us to understand the potential harms associated with the modRNA pro-vaccines in a way that begins to restore the public trust in the scientific process and health institutions, by engaging in a process of introspection and improvement of regulatory processes and decision-making.
These expedited and still experimental vaccines are the most complicated medical products ever deployed. Pfizer’s recently retired head of vaccine research, Dr. Kathrin Jansen, was quoted in Nature as saying ““We flew the aeroplane while we were still building it.” (223)
It is now time to ground the plane pending the answers to these, and no doubt, many more questions.
.
17. Topics for further discussion
The following are topics for further discussion:
· Compliance with pharmacovigilance standards
· DNA-RNA hybridization
· Developmental and Reproductive Toxicology (DART)
· Studies examining the toxicity of N1-methylpseudouridine
· Studies examining the effects of UTRs (untranslated regions) in the modRNA, particularly those derived from humans, and those that have mitochondria-related sequences.
· The effects of differential glycosylation patterns on the pharmacology and toxicology of the spike protein.
· The consequences of RNA fragmentation and the possibility of aberrant protein production.
· Endotoxin and sequestration by LNPs
18. Consolidated list of questions
Question 1: Please confirm that neither label includes the indication that the vaccine reduces “the risk of death, hospitalization and serious illness” of COVID-19 disease.......................................................................................................................................................... 5 Q1
Question 2: Has FDA reviewed data that meet the standard of “substantial evidence” “consisting of adequate and well-controlled investigations” and support the claim that either vaccine reduces “the risk of death, hospitalization and serious illness” of COVID-19 disease. Please provide.5 Q2
Question 3: If FDA considers the data it has reviewed as supporting the claim that either vaccine reduces “the risk of death, hospitalization and serious illness” of COVID-19 disease and meeting the standard of “substantial evidence” “consisting of adequate and well-controlled investigations,” please state whether they do so according to the evidentiary standards set forth in FDA’s 1998 guidance (19) or or according to later 2019 (20) and 2023 (21) documents that expand the scope of the types of data that can be used to support certain claims or expanded indications............................ 6 Q3
Question 4: If FDA has reviewed data that do not meet the standard of “substantial evidence” “consisting of adequate and well-controlled investigations” but purports to support this claim, please provide the data and state the evidentiary standard such data meet, if at all.6 Q4
Question 5: If FDA has reviewed data that do not meet the standard of “substantial evidence” “consisting of adequate and well-controlled investigations” please describe the deficiencies in the data that preclude the inclusion of language asserting that the COVID-19 vaccines reduce “the risk of death, hospitalization and serious illness” of COVID-19 disease............................................................................................................... 6 Q5
Question 6: Please advise whether, in the absence of data meeting the “substantial evidence” standard as well as authorization by FDA of a labeling change to include a claim that the vaccine reduces “the risk of death, hospitalization and serious illness” of COVID-19 disease, manufacturers making such a claim would be in violation of statues and regulations regarding off-label promotion? How is your answer influenced by FDA’s draft guidance on Scientific Information on Unapproved Uses (SIUU)? (22)....................................................................................................................... 6 Q6
Question 7: What is the regulatory status of Dr. Marks’ (4) statement regarding a “dramatic reduction in the risk of death, hospitalization and serious illness afforded by the vaccines”? Coming from Dr. Marks, a senior FDA official, does this represent an amendment to the approved labeling, a regulatory guidance, an enforcement policy, SIUU, or personal medical opinion?................................................................................... 6 Q7
Question 8: Although this draft guidance applies to sponsors, given Dr. Marks’ statement concerning a “dramatic reduction in the risk of death, hospitalization and serious illness afforded by the vaccines,” and the absence of the corresponding claim in the package insert, is it FDA’s intent that the modRNA COVID-19 vaccines be used to reduce these serious outcomes?...................................................................................... 6 Q8
Question 9: Per Question 8, if it is FDA’s intent that the COVID-19 vaccines be used to reduce serious outcomes, would the issuance of such a statement on this use by FDA without the qualifying language concerning this unapproved use, misleadingly imply that this use had been approved by FDA?6 Q9
Question 10: Did the provision of EUA product to patients who were counseled that these products reduce the risk of death or serious outcomes, violate the provider agreement, which requires that the provider must confine representations to those consistent with the contents of the Fact Sheet - eg (23, 24) ?6 Q10
Question 11: Please confirm that the above excerpts do appear in the respective package inserts...................... 7 Q11
Question 12: Has FDA reviewed data that meet the standard of “substantial evidence” “consisting of adequate and well-controlled investigations” and support the claim that either vaccine is safe and effective for use in pregnancy or lactation. Please provide..................... 7 Q12
Question 13: If FDA considers the data it has reviewed as supporting the claim that either modRNA vaccine is safe and effective for use in pregnancy and meeting the standard of “substantial evidence” “consisting of adequate and well-controlled investigations,” please state whether they do so according to the evidentiary standards set forth in FDA’s 1998 guidance (19) or according to later 2019 (20) and 2023 (21) documents that expand the scope of the types of data that can be used to support certain claims or expanded indications................................................................... 7 Q13
Question 14: If FDA has reviewed data that do not meet the standard of “substantial evidence” “consisting of adequate and well-controlled investigations” but purports to support this claim, please provide the data and state the evidentiary standard such data meet, if at all.7 Q14
Question 15: If FDA has reviewed data that do not meet the standard of “substantial evidence” “consisting of adequate and well-controlled investigations” please describe the deficiencies in the data that preclude the removal of tempering labeling language and/or the inclusion of language asserting that the COVID-19 vaccines are safe and effective in pregnancy and lactation............................................................................... 7 Q15
Question 16: Please advise whether, in the absence of data meeting the “substantial evidence” standard as well as authorization by FDA of a labeling change to include a claim that the vaccine is safe and effective in pregnancy, manufacturers making such a claim would be in violation of statues and regulations regarding off-label promotion?........................................................................................................................................ 7 Q16
Question 17: Please confirm that CDC’s recommendation for use of the COVID-19 vaccines in pregnancy and lactation, along with CDC’s representation that “Evidence shows that: COVID-19 vaccination during pregnancy is safe and effective,” is misleadingly inconsistent with the wording in the COMIRNATY and SPIKEVAX package inserts concerning insufficient data to inform vaccine-associated risks in pregnancy, whether the vaccines are excreted in breast milk, and the lack of data on the effects of the vaccines on the breastfed infant or on milk production/excretion.................. 7 Q17
Question 18: Please confirm that the absence of prominent language detailing the risks of these products in pregnancy and lactation as described in FDA approved labeling, from CDC’s related recommendations, exacerbates the misleading nature of these recommendations.8 Q18
Question 19: Please provide the contents and URLs of all current FDA and CDC web pages that discuss the use of these products in pregnancy and lactation. Please detail what steps will be taken to ensure that prominent language will be placed, if currently absent, detailing the risks of these products in pregnancy and lactation as described in FDA approved labeling........................................................................................ 8 Q19
Question 20: Please confirm that FDA’s endorsement of CDC’s recommendation for use of the COVID-19 vaccines in pregnancy and lactation, along with CDC’s related representations described above is misleadingly inconsistent with the wording in the COMIRNATY and SPIKEVAX package inserts concerning insufficient data to inform vaccine-associated risks in pregnancy, whether the vaccines are excreted in breast milk, and the lack of data on the effects of the vaccines on the breastfed infant or on milk production/excretion........................................................................ 9 Q20
Question 21: Please confirm that the absence of prominent language detailing the risks of these products in pregnancy and lactation as described in FDA approved labeling, from FDA’s written and video endorsement of CDC’s related recommendations, exacerbates the misleading nature of both FDA’s endorsement and CDC’s recommendations..................................................................................................... 9 Q21
Question 22: Did the provision of EUA product to patients who were counseled that these products were safe and effective for use in pregnancy and lactation violate the provider agreement, which requires that the provider must confine representations to those consistent with the contents of the Fact Sheet - eg (23, 24) ....................................................................................................................................................... 9 Q22
Question 23: Did the Secretary of HHS authorize deviations from cGMP regarding the COVID-19 vaccines? Was this a general waiver for all COVID-19 vaccines or for specific vaccines and/or specific issues? Did such a waiver cover any cGMP issues stemming from the Process 1 to Process 2 change for Pfizer’s product?Please provide a copy of all relevant cGMP waivers............................................................ 11 Q23
Question 24: When did FDA first learn that Pfizer would be changing from Process 1 to Process 2?.................. 11 Q24
Question 25: After learning about Pfizer’s process change, did FDA consider this change to constitute, absent a comparability analysis, grounds for a non-approvable status or the issuance of something akin to the EMA Major Objection?........................................... 11 Q25
Question 26: To what extent did the challenges related to Pfizer’s process change contribute to FDA’s change in regulatory approach from a BLA pathway described in the June 2020 guidance (32) to an EUA pathway describedthe October 2020 guidance (33)?...... 11 Q26
Question 27: Within the BLA framework described in the June 2020 guidance (32) what comparability requirements did FDA place or would have placed on Pfizer related to the proposed change in manufacturing process? How would these requirements differ under an EUA framework?11 Q27
Question 28: What combination of provisions, akin to those adopted by EMA, such as GMP waivers, additional pre-EUA data provided by Pfizer, post-EUA obligations and commitments, did FDA make in order to obviate any delays in authorization or approval caused by the process change?12 Q28
Question 29: Absent these provisions, by how long would the issuance of Pfizer’s EUA have been delayed?..... 12 Q29
Question 30: Relating to this process change, did FDA request a risk assessment from Prizer? Was one provided and when? Did FDA conduct its own risk assessment? Was any risk assessment addressing this issue, if one exists, disclosed to VRBPAC or publicly. If not please provide.12 Q30
Question 31: Please confirm that there is no reference to the Process 1 to Process 2 manufacturing change in the meeting materials for the VRBPAC meeting of December 10 2020. Was VRBPAC fully informed of the fact and details of the manufacturing change, including protocol Amendment 7, and if so when and in what form?.................................................................................................................................................. 12 Q31
Question 32: What was the regulatory basis for authorizing a process change based on a descriptive comparability analysis involving 250 subjects per arm? Does this analysis meet the BLA “substantial evidence” or merely the EUA “totality of evidence standard”? If the answer is the latter, how is this lowered standard consistent with FDA’s representation in its October 6 2020 (33) guidance and to VRBPAC on October 22, 2020 (38) that it would still require data “from at least one well-designed Phase 3 clinical trial that demonstrates the vaccine’s safety and efficacy in a clear and compelling manner”?12 Q32
Question 33: Regarding the process change, was VRBPAC fully informed and educated about the lowering of the “substantial evidence” or “clear and compelling” standards to a “totality of evidence” standard? When? In what form?............................................. 12 Q33
Question 34: Was VRBPAC fully informed and educated about the existence and details of any cGMP waivers? When and in what form? Other than the publication of Pfizer’s study and protocol in the NEJM on the same day as the VRBPAC meeting, did VRBPAC members receive these materials prior to the December 10 2020 meeting?........................................................................................................................ 12 Q34
Question 35: How many different lots of Process 2 Drug Product (DP) were deployed in Pfizer’s pivotal trial C4591001? How many subjects received Process 2 DP (by lot number)?..................................................................................................................................... 13 Q35
Question 36: Please confirm the information provided by MHRA in their FOI response 23/510 that the first subject to receive Process 2 DP did so on October 18 2020. Please provide the date when the last subject to receive Process 2 DP did so........................................ 13 Q36
Question 37: Please confirm the information provided in EMA’s report (30) that the descriptive clinical comparability analysis was expected in February 2021. If this was not the case, what was the timeline for submission to FDA of Pfizer’s descriptive clinical comparability analysis?13 Q37
Question 38: What was the regulatory basis for issuing Pfizer’s EUA in the absence of this analysis?................ 13 Q38
Question 39: What actions did FDA take when Pfizer failed to submit its descriptive clinical comparability analysis by the specified date?13 Q39
Question 40: What was the regulatory basis for re-issuing Pfizer’s EUA with its various amendments including those involving booster shots and new variant versions in the absence of this analysis?13 Q40
Question 41: Please confirm the information provided by MHRA in their FOI response 23/510 that this analysis was never conducted and submitted to FDA.................................................................................................................................................................... 13 Q41
Question 42: Please confirm the information provided by MHRA in their FOI response 23/510 that analysis was removed from the protocol in amendment 20 in September 2022.......................................................................................................................................... 13 Q42
Question 43: What was the justification provided by Pfizer for not conducting or submitting this analysis? Please confirm that all or part of this justification is similar to that provided by MHRA in their FOI response 23/510 that this was “due to the extensive usage of vaccines manufactured via “Process 2.”13 Q43
Question 44: Comparing and contrasting with Question 32 and noting FDA’s 1998 (19), 2019 (20), and 2023 (21) guidance documents regarding evidentiary standards for clinical data, what is the regulatory basis for authorizing or approving a vaccine based on only one clinical study of DP made by a process that differs with DP currently used and made by a process for which there is no “substantial evidence” of clinical comparability “consisting of adequate and well-controlled investigations.”............................................................................................................................. 13 Q44
Question 45: If FDA is relying on “extensive usage” in a manner apparently similar to MHRA, is this intended to constitute Real World Evidence (RWE) that can support approvals under some circumstances only described in FDA’s September 19 guidance? (21) Has this RWE been subject to the appropriate controls described in a guidance only recently (Aug 30, 2023)? (41)............................................................................ 13 Q45
Question 46: Was Process 2 DP used in any of Pfizer’s other trials or sub-trials? If so, which?......................... 13 Q46
Question 47: Did FDA express any concern to Pfizer about any of the process-related issues identified above, including the poly(A) tail pattern. the 5’ cap, mRNA integrity, dsRNA, the pattern and identity of RNA and truncated or fragmented RNA, and the identity and molecular weights of proteins expressed after modRNA transfection? How were these concerns resolved? What was the timeline of this process from FDA’s first awareness, to FDA’s expression of concern or questions, to Pfizer’s response and to resolution?...................................................................................... 14 Q47
Question 48: Were there concerns similar to those listed in Question 47 regarding the Moderna product? Please describe.14 Q48
Question 49: Was FDA aware of the concerns expressed by EMA or other regulatory agencies on the subjects discussed in Question 47 and the actions they took to address them? When? Was there any consultation or coordination between agencies?......................... 14 Q49
Question 50: Did FDA express any concern to Pfizer about any issue related to residual DNA such as the robustness of the DNase digestion step. How were these concerns resolved? What was the timeline of this process from FDA’s first awareness, to FDA’s expression of concern or questions, to Pfizer’s response and to resolution?......................................................................................................................................... 16 Q50
Question 51: How is the continuing concern well into at least 2022 about residual DNA consistent with the issuance of COMIRNATY’s BLA in August 2021 and the authorization of children’s doses in October 2021?................................................................................... 16 Q51
Question 52: Were there concerns similar to those listed in Question 50 regarding the Moderna product? Please describe.16 Q52
Question 53: Was FDA aware of the concerns expressed by EMA or other regulatory agencies on the subjects discussed in Question 50 and the actions they took to address them? When? Was there any consultation or coordination between agencies?......................... 16 Q53
Question 54: Is FDA aware that “GMP-like” may have been used in the manufacture of Pfizer mRNA?............. 17 Q54
Question 55: For which phases of Pfizer COVID-19 vaccine preclinical, clinical and post EUA use was “GMP-like” plasmid used?17 Q55
Question 56: What was the source of Pfizer’s plasmid and its GMP provenance (i.e. GMP, or GMP-like)? Was this obtained from Pfizer’s Gene Therapy Division at the large-scale pDNA manufacturing facility in Chesterfield, MO? Was FDA aware of the source of plasmid?. 17 Q56
Question 57: How does “GMP-like” plasmid differ from GMP-compliant plasmid?............................................. 17 Q57
Question 58: Regarding the COVID-19 mRNA vaccines, for what other preclinical, clinical and post EUA purposes were processes or materials “GMP-like” rather than “GMP compliant? Were these instances of “GMP-like” a reflection of EUA regulations or FDA’s non-enforcement of GMP requirements?17 Q58
Question 59: Where there any instances of “GMP-like” processes or materials related to the development or manufacturing of the Moderna COVID-19 mRNA vaccine?...................................................................................................................................................... 17 Q59
Question 60: For the Pfizer product, which process was used to make the drug product tested in the non-clinical pharmacology and toxicology studies described in the Summary Basis for Regulatory Action. (47) Was test article taken from clinical or commercial scale production material, or from especially conducted non-clinical runs?................................................................................................................................................ 18 Q60
Question 61: For the Pfizer product, which non-clinical studies were performed with the V8 version and which with the V9 version? Please confirm that the type made by both Processes 1 and 2 was the V9 type.......................................................................................... 18 Q61
Question 62: For both the Pfizer and Moderna products, please summarize and tabulate differences in the composition of Drug Product used in non-clinical, clinical, and post-authorization COVID-10 vaccines, paying particular attention to the modRNA ORF sequence, sequence of non-coding regions, extent and type of nucleoside medication, pattern of codon optimization, LNP and buffer composition. Please indicate the reason for each change and what analytical, non-clinical or clinical comparability studies were performed to qualify these changes............................................. 18 Q62
Question 63: For both the Pfizer and Moderna products, please summarize and tabulate all manufacturing changes from the formulation and process used to produce non-clinical test material to currently produced vaccine that may have changed the amount, type of size distribution of DNA or RNA in the final DP, the amount and type of impurities, as well as critical quality attributes and properties of the LNPs. Please indicate the reason for each change and what analytical, non-clinical or clinical comparability studies were performed to qualify these changes...................................... 18 Q63
Question 64: In light of the above review of basic cell biology, and FDA’s “first principle” premise of nuclear membrane inviolability, what studies have FDA conducted or solicited or will FDA be conducting or soliciting from Pfizer and Moderna regarding the intracellular kinetics of residual DNA?20 Q64
Question 65: In light of the above attestations as to the risks of insertional mutagenesis, would FDA revise Dr. Marks’ earlier statement concerning the plausibility of risk of chromosomal integration of residual DNA?....................................................................... 22 Q65
Question 66: As with conventional pharmacokinetics (PK) (see 0Error! Reference source not found.), a full understanding of the cellular kinetics of any drug is essential to understand its pharmacology and toxicology and is not an academic nicety. What studies will FDA be conducting on its own, or soliciting from Pfizer or Moderna, regarding the intracellular kinetics of modRNA?.......................................................................... 23 Q66
Question 67: In light of the above evidence, what studies is FDA conducting in its own labs, is aware of being undertaken by other government agencies, or is soliciting from Pfizer of Moderna to characterize the reverse transcription of vaccinal modRNA to DNA?........... 24 Q67
Question 68: In light of the above evidence, what assessments have FDA conducted, solicited from Pfizer of Moderna or received from elsewhere to characterize the risks of reverse transcription of vaccinal modRNA to DNA?.................................................... 24 Q68
Question 69: What studies have FDA requested from Pfizer or Moderna to determine whether genomic insertion may occur with residual DNA or from reverse transcribed vaccinal modRNA after modRNA vaccine administration?............................................................. 25 Q69
Question 70: What in vitro or in vivo models does FDA consider suitable to assess genomic integration of residual DNA, after appropriate validation?25 Q70
Question 71: What studies have FDA conducted, or will conduct to determine whether genomic insertion may occur with residual DNA or from reverse transcribed vaccinal modRNA? Please provide details.................................................................................... 25 Q71
Question 72: Have FDA conducted studies using the models described in or adapted from Sheng-Fowler et al (96-98) to assess integration or oncogenesis after administration of the oncogene expression plasmids within the same or similar LNPs used in the Pfizer or Moderna COVID-19 vaccines? Please provide details.................................................................................................................................................................... 25 Q72
Question 73: Have FDA conducted studies using the models described by or adapted from Sheng-Fowler et al (96-98) to assess integration or oncogenesis after co-administration of the oncogene expression plasmids and sequence elements from the plasmid vectors used for modRNA COVID-19 vaccine production? Please provide details................................................................................................................ 25 Q73
Question 74: Have FDA conducted studies using the models described by or adapted from Sheng-Fowler et al (96-98) to assess integration or oncogenesis after co-administration of the oncogene expression plasmids and sequence elements from the plasmid vectors used for modRNA COVID-19 vaccine production within same or similar LNPs used I the COVID-19 vaccines? Please provide details......................................... 25 Q74
Question 75: What studies have FDA conducted, will conduct or have solicited from Pfizer or Moderna, to determine whether extrachromosomal expression or transmission of residual DNA occurs, and to determine the attendant risks, if detected?................................... 26 Q75
Question 76: Given that these guidelines did not contemplate the highly efficient transfection of nucleic acid by LNPs (see 6.2), please provide a justification as to why FDA’s original (pre-2007) recommendation to conduct preclinical studies to assess vaccine-induced autoimmune disease should not be reinstated?27 Q76
Question 77: Has FDA conducted or solicited from Pfizer or Moderna a risk assessment related to vaccine -associated autoimmune disease?27 Q77
Question 78: Has FDA communicated with other US government agencies such as NIH or CDC about the risk of modRNA vaccine -associated autoimmune disease?...................................................................................................................................................... 27 Q78
Question 79: Is FDA aware of risk assessments or studies performed by other US government agencies such as NIH or CDC related to the risk of modRNA vaccine -associated autoimmune disease? What is the nature of this work?..................................................... 27 Q79
Question 80: What studies or risk assessments has FDA conducted or will conduct, has solicited, or will solicit from Pfizer or Moderna, to determine the contribution of non-integrating mechanisms of toxicity of DNA to the overall safety profile of the modRNA vaccines?27 Q80
Question 81: What lessons regarding DNA toxicity can learned from the viral vector COVID-19 vaccines (Janssen, Astra-Zeneca) and applied to the toxicity of residual or reverse transcribed DNA associated with the modRNA vaccines?................................................... 27 Q81
Question 82: Why does FDA consider the ratio of residual DNA to the amount of RNA relevant in determining the absolute risk of residual DNA in modRNA vaccines? Is this ratio used in the setting of specifications for Drug Substance or Drug Product? What is this specification?29 Q82
Question 83: What is FDA’s estimate of the fold-increase of transfection for nucleic acid achieved by the LNPs used in the Pfizer and Moderna modRNA COVID-19 vaccine formulations?.................................................................................................................. 29 Q83
Question 84: Per Question 83, Is this estimate based on FDA’s own studies? If so please describe those studies? If not, was this based on data provided by Pfizer and Moderna? Please provide details................................................................................................... 29 Q84
Question 85: If FDA permits an upward adjustment in the residual DNA limit in a case where less risk is perceived (i.e. oral dosing), what is FDA’s rationale for not downwardly adjusting the residual DNA limit, in cases where there is more reason to be concerned (i.e. enhanced transfection using LNPs)?29 Q85
Question 86: What animal or human studies has FDA conducted or solicited from Pfizer or Moderna concerning the biodistribution of residual DNA, quantified in terms of number of copies? Please provide.................................................................................................... 30 Q86
Question 87: What algorithms does FDA use to compute integration risk based on the number of copies and sizes of DNA fragments, their distribution and persistence? Please provide details and a record of the calculations performed,.............................................. 30 Q87
Question 88: How does FDA characterize any possible integration risk for the purposes of determining “safe” exposure levels? For example, does FDA consider exposure to integration-competent DNA capable of producing an (mostly) irreversible effect similar to exposure to ionizing radiation, or rather as an exposure to a toxin that produces a concentration dependent reversible effect?................................................................. 30 Q88
Question 89: What algorithm does FDA use to adjust the limit of residual DNA per dose, based on FDA’s characterization of risk (per Question 88), the pharmacokinetic properties of residual DNA within LNPs, the interval between multiple doses of COVID-19 vaccine, the interval and dose between the administration of conventional DNA-containing vaccines or non-COVID-19 modRNA vaccines that may be introduced in the future?30 Q89
Question 90: Please describe the method used to adjust raw estimates of residual DNA for amplicon length and amplification efficiency.30 Q90
Question 91: Please confirm which test methods are used to determine RNA and residual DNA in Drug Substance and Drug Product.31 Q91
Question 92: Please provide a justification for why UV or fluorescence methods have not been used to determine the amount of residual DNA, as they appear to be used to estimate RNA.............................................................................................................................. 31 Q92
Question 93: What are the sequences and lengths of amplicons used in the “validated quantitative PCR assay” you refer to that is used to estimate the amount of residual DNA?.......................................................................................................................................... 31 Q93
Question 94: For both Moderna and Pfizer, what is the smallest length of DNA that can be detected by the particular amplicons used, and under the assay conditions used, for the “validated quantitative PCR assay” used to estimate residual DNA?............................. 31 Q94
Question 95: What studies have FDA performed or solicited from Pfizer or Moderna to characterize the size distribution of residual DNA fragments as a function of amplicon length? Please provide............................................................................................................... 31 Q95
Question 96: What is the percentage of total residual DNA detected by qPCR?................................................ 31 Q96
Question 97: Please supply the results of residual DNA assay in DS, or DP, for all lots of Pfizer-BionTech or Moderna EUA or BLA COVID-19 vaccines. Please provide the total number of doses supplied, and if known, administered, of each lot, within the USA. Please supply the date of first release for each lot.31 Q97
Question 98: What measures have been taken to reduce the level of residual DNA contamination?.................. 31 Q98
Question 99: What studies have FDA performed or solicited from Pfizer or Moderna to characterize the differences between qPCR and UV or fluorescence methods of estimating the amount of residual DNA?....................................................................................... 31 Q99
Question 100: Please supply the test protocols for estimating DNA or RNA by qPCR, UV absorption or fluorescence methods, including details of sample preparation to ensure recovery from LNPs and the reduction of confounding of RNA measurements by DNA, or vice-versa.31 Q100
Question 101: Please confirm that residual DNA is measured at the end of the IVT process, and not in the final drug product. Please justify why it should is not also measured in the final DP formulation, commenting on whether there is free DNA outside of the LNP.......... 31 Q101
Question 102: Are residual DNA fragment size or size distribution critical quality attributes for modRNA DS or DP? What methods are used to determine fragment size and distribution?...................................................................................................................... 32 Q102
Question 103: Are residual DNA fragment size or size distribution critical quality attributes included in release specifications?32 Q103
Question 104: Are residual DNA fragment size or size distribution determined as part of the lot release requirements?32 Q104
Question 105: According to a FOIA disclosure from Health Canada p24/584 in (118), Pfizer claimed they had never been asked by any regulator to conduct a DNA fragment size distribution analysis. Please confirm. If true, please justify.................................................. 32 Q105
Question 106: Did Pfizer provide a DNA fragment size distribution analysis? Please provide. Otherwise please explain why they were not asked to do so.33 Q106
Question 107: Did Moderna provide a DNA fragment size distribution analysis? Please provide. Otherwise please explain why they were not asked to do so.................................................................................................................................................................... 33 Q107
Question 108: What studies has FDA conducted or solicited from Pfizer or Moderna to describe the fragment size distribution of residual DNA in modRNA vaccines? Please provide methodological details........................................................................................... 33 Q108
Question 109: What percentage of lots of COVID-19 modRNA vaccines failed release testing either by manufacturers or FDA because fragment size criteria were out of specification?............................................................................................................................. 33 Q109
Question 110: If fragment size data were not part of release criteria, but nonetheless measured, what percentage of released lots of COVID-19 modRNA vaccine contained fragments larger than 200 bp? Please stratify by manufacturer, presentation (adult vs. children’s dose etc.), and variant type (original, bivalent, XBB.1.5)........................................................................................................................................ 33 Q110
Question 111: Given the finding that released lots did contain fragments of residual DNA greater than 200 bp and given the above statement in the cited WHO 2007 (115) document, what adjustments to the 10 ng dose limit are required to preserve the same safety margin?33 Q111
Question 112: What studies has FDA conducted or solicited from Pfizer or Moderna to determine the prevalence of intact sequence elements from the plasmid vectors in the pool of residual DNA found in COVID-19 modRNA vaccines?.................................................... 33 Q112
Question 113: Please summarize and tabulate all changes to the sequences of the DNA plasmid vector and the modRNA DS used in the preclinical tests, clinical studies, and post-authorization to the current versions of COVID-19 vaccines. Please indicate the reason for each change and what analytical, non-clinical or clinical comparability studies were performed to qualify these changes............................................. 35 Q113
Question 114: Per Question 113, if no preclinical or clinical studies were performed for any given change, please provide a rationale.35 Q114
Question 115: What risk-assessment or studies have FDA conducted or solicited from Pfizer or Moderna related to the transfection of an antibiotic resistance gene within residual DNA into a vaccinee?..................................................................................................... 35 Q115
Question 116: What risk-assessment or other studies have FDA conducted or solicited from Pfizer or Moderna related to the transfection of antibiotic resistance genes into commensal or infection pools of bacteria in a vaccinee?................................................................. 35 Q116
Question 117: What risk-assessment or studies have FDA conducted or solicited from Pfizer or Moderna related to the transfection of antibiotic resistance genes into commensal or infection pools of bacteria in a vaccinee?................................................................. 35 Q117
Question 118: What risk-assessment or studies have FDA conducted or solicited from Pfizer or Moderna related to the transfection of antibiotic resistance genes into environmental (e.g. soil, wastewater) bacteria?.............................................................................. 35 Q118
Question 119: Please provide details of the “in-house mRNA” used by Wang et al., (11) particularly its source and similarity to EUA of BLA material. Please provide all raw data for this study, and describe the involvement of FDA staff and their relationship to the student. Please provide the protocols or other documentation likely needed for submission to the R&D committees that would have been needed to approve the conduct of the study.35 Q119
Question 120: Please confirm that the plasmid template used to produce all Pfizer-BioNTech COVID-19 vaccines made under EUA or BLA, to date (including the XBB.1.15 vaccine) contain sequences for 1) SV40-promoter-enhancer-ori, 2) SV40 poly(A) signal; 3) HSV poly(A) signal.38 Q120
Question 121: Please describe FDA’s expectation, by statute, regulation, or practice for sponsors to disclosure all sequence elements contained in the plasmid template used for the production of modRNA or mRNA vaccines..................................................................... 38 Q121
Question 122: Please describe whether Pfizer disclosed the full plasmid sequence of its plasmid to FDA and whether this disclosure included specific details of sequence elements, including the three sequences listed above apparently not disclosed to EMA or Health Canada. Please describe whether these disclosures included an annotated plasmid map. Please answer this question for all variant (Wuhan, bivalent, XBB1.5) vaccine versions, whether under EUA or BLA. Please provide the dates of disclosure of the full sequence and the details of any sequence elements not disclosed along with the full sequence.38 Q122
Question 123: If these three sequence elements were not detailed at the same time as the full sequence, please provide Pfizer’s justification for not doing so.................................................................................................................................................................... 38 Q123
Question 124: Please provide the date when FDA asked Pfizer whether or these sequences were present in their plasmid.38 Q124
Question 125: Please describe whether and when Pfizer disclosed to FDA the function of these three sequences.38 Q125
Question 126: Please state when FDA asked Pfizer to describe the function of these sequences. Please describe the function of these three sequences.38 Q126
Question 127: Please state whether Pfizer or FDA provided or performed a risk assessment related to the presence of these sequences, as intact sequences in residual DNA in Drug Product? If one has been submitted or prepared, please provide a copy.......................... 38 Q127
Question 128: Per Question 127, does this assessment consider the actions of the SV40 enhancer-promoter-ori described in section 7.2.2? If not, please discuss these topics..................................................................................................................................... 38 Q128
Question 129: Please state when FDA asked Pfizer to provide a risk assessment related to these sequences... 38 Q129
Question 130: If, according to the FDA 2010 guidance (63) the risks of DNA can be lessened by reducing the amount of residual DNA, please provide a justification for increasing the load of DNA by the inclusion of SV40 sequences that are, according to EMA, (116) “non-functional.”38 Q130
Question 131: If, per the above questions, Pfizer failed to make the appropriate disclosures regarding the presence, function or assessment of risk of these sequences in a timely fashion, what regulatory actions were and will be taken against Pfizer? What was Pfizer’s justification for failing to make these disclosures?................................................................................................................................................ 39 Q131
Question 132: If the SV40 sequences are indeed non-functional,” and their inclusion not unavoidable, it would appear that intact or fragmented SV40 or HSV sequences found in residual DNA constitute “extraneous material.” What investigative or enforcement actions has FDA taken to correct this apparent violation of the regulations that ““Products shall be free of extraneous material.”............................................................... 39 Q132
Question 133: Did FDA receive a document similar to that provided to Health Canada? (126) When? Please supply unredacted text.39 Q133
Question 134: What evidence did Pfizer present to justify the statement: “residual DNA is expected to degrade rapidly “ Did FDA ask Pfizer to provide this evidence? Please provide............................................................................................................................. 39 Q134
Question 135: What evidence did Pfizer present to justify the statement: “residual DNA […] has a very low likelihood of reaching the nucleus. “ Did FDA ask Pfizer to provide this evidence? Please provide.............................................................................................. 39 Q135
Question 136: Did Pfizer quantify, with justification, just how likely or unlikely the sequences described could reach the nucleus? Did FDA ask Pfizer to provide this evidence? Please provide....................................................................................................................... 39 Q136
Question 137: Given the absence of a nuclear member in mitosis ( 4.2), and the ability of the SV40 sequence to act as a nuclear localization signal, (69, 127) did FDA challenge Pfizer on the assertion that “residual DNA […] has a very low likelihood of reaching the nucleus.“Please provide.39 Q137
Question 138: Did Pfizer quantify, the likelihood of expression of the resistance gene, as well as the duration of its “transience.” Did Pfizer describe what biological this gene would have if expressed and explain why this would not pose a safety risk? Did FDA seek answers to these questions?39 Q138
Question 139: Has FDA asked Pfizer to remove the SV40 sequences from their plasmids? What is the schedule for this? Ny what regulatory pathway will these non-SV40 versions of Pfizer’s pro-vaccines be approved? Will RCT’s be required?.......................................... 39 Q139
Question 140: .What investigations were performed by Pfizer, Moderna, FDA, or other government to determine the presence of “unexpected open reading frames” or “unintended sequences of biological significance” in both strands of the plasmid vector used to produce the modRNA COVID-19 vaccines?40 Q140
Question 141: Please provide the study reports of any investigations performed per Question 140, along with risk assessments related to the findings.40 Q141
Question 142: When did FDA become aware that lipids may form adducts with nucleic acids?.......................... 41 Q142
Question 143: What is the nature of the lipid-RNA species and why might they be a concern?.......................... 41 Q143
Question 144: Did FDA have a similar concern for lipid-RNA species as did EMA? Were these concerns based on formation of aldehyde-related adducts, or other mechanisms?...................................................................................................................................... 41 Q144
Question 145: How was this concern lipid-RNA species resolved?.................................................................. 41 Q145
Question 146: Given that the work on the lipid-RNA species was to be provided by January 1 2021, when exactly did this occur?41 Q146
Question 147: If lipid-RNA species prior to resolution of this issue, how many doses of mRNA-1273 had been administered either in clinical trials or post approval/ authorization?............................................................................................................................... 41 Q147
Question 148: Are there specific guidelines and limits on these adducts? How are they controlled? Did Pfizer and Moderna comply with these guidelines?41 Q148
Question 149: Given what was known at the time about lipid adducts and their possible biological consequences, what studies analytical, preclinical or clinical studies did FDA require from Pfizer when they changed their buffer? What were the results or requested or voluntarily provided studies?42 Q149
Question 150: Why were the possible biological consequences of a buffer change fully disclosed to VRBPAC who were being asked to make recommendations based on the totality of scientific evidence available and a consideration of known and potential risks?.............. 42 Q150
Question 151: By way of tabulation, please compare and contrast, the processes used to produce the original monovalent version of the COVID-19 modRNA provaccines, and the bivalent. Please provide separate comparisons or Moderna and Pfizer............................ 43 Q151
Question 152: Please provide the questions asked by FDA and the justifications provided by Moderna and Pfizer to support the claim of manufacturing comparability............................................................................................................................................... 43 Q152
Question 153: Were Moderna and Pfizer asked to provide an assessment of toxicological equivalency of the heterotrimer spike proteins to their homotrimer counterparts? Please provide........................................................................................................................ 43 Q153
Question 154: Were Moderna and Pfizer asked to conduct in vitro, animal or clinical comparability testing, particularly to demonstrate toxicological equivalency of the heterotrimer spike proteins to their homotrimer counterparts? Please provide......................................... 44 Q154
Question 155: Which peer reviewed paper(s) or regulatory document(s), including submissions from Pfizer or Moderna describe the details of “animal studies with the mRNA delivery technology done over the past decade” that “show no evidence of genotoxicity.” Please provide.44 Q155
Question 156: Given that, according to a paper co-authored by a founder of Moderna, (67) LNP particle size is a major determinant of distribution, and also according to FDA, “because biodistribution and retention is a property of the LNP rather than the mRNA,” how does this the study support“the approval of SPIKEVAX BLA”?........................................................................................................................................ 50 Q156
Question 157: Following from Question 156, Given that, according to a paper co-authored by a founder of Moderna, (67) LNP particle size is a major determinant of distribution, and also according to FDA, “because biodistribution and retention is a property of the LNP rather than the mRNA,” how does this the study support the authorization of Pfizer product using Tris buffer?.................................................................. 50 Q157
Question 158: Given that the composition of mRNA 1647 is critical to understand the relevance of any studies that are used to support the authorization or approval of mRNA 1273, please provide the full formulation details of mRNA 1647, such as those redacted from the distribution study report. (131)50 Q158
Question 159: Per Question 158, did the formulation of mRNA 1647 used in Moderna’s distribution study contain Tris?50 Q159
Question 160: Given the manufacturing controls alluded to by Moderna (129) to reduce lipid adduct formation, when, relative to the conduct of the Moderna’s distribution study, were these controls implemented?..................................................................................... 50 Q160
Question 161: Please confirm the accuracy of FDA’s document (151) in stating the that the product used in Moderna’s distribution study was “manufactured using the same procedure as SPIKEVAX”?.................................................................................................... 50 Q161
Question 162: What consideration has FDA made concerning the selection of suitable animal models for pharmacology, biodistribution and other safety studies for modRNA products encoding antigens whose interaction with host ligands may be species specific?............. 51 Q162
Question 163: What data or literature were provided by Pfizer to support their expectations (160) regarding the degradation or mRNA or spike protein?53 Q163
Question 164: Given emerging data suggesting vaccinal modRNA persistence for significantly longer (72, 73, 75, 78) than the “short time” described in the WHO guideline on mRNA vaccines,(71) and given FDA’s participation in the drafting of that document, what revisions has FDA proposed or will propose to that document?................................................................................................................................................... 53 Q164
Question 165: Since the introduction of the modRNA COVID-19 vaccines, to what extent did FDA agree with CDC messaging suggesting that the modRNA is eliminated “within a few days” and the spike protein “within a few weeks” (see 11.1)?....................................... 53 Q165
Question 166: Given emerging data suggesting vaccinal modRNA persistence for significantly longer (72, 73, 75, 78) than the “few days” and spike protein persistence for significantly longer than “a few weeks”(74, 75, 78, 163) (see 11.1) what revisions has FDA proposed or will propose to CDC or other government entities to correct the earlier statements?....................................................................................................... 53 Q166
Question 167: Since the introduction of the modRNA COVID-19 vaccines and given data generated by Pfizer and Moderna in animals showing a wide distribution of LNPs and/or modRNA, to what extent did FDA agree with CDC messaging suggesting that the modRNA vaccines stay at the site of injection, where they act (see 11.1)?............................................................................................................................ 54 Q167
Question 168: To what extent does FDA now agree with CDC messaging suggesting that the modRNA vaccines stay at the site of injection, where they act (see 11.1)?.......................................................................................................................................................... 54 Q168
Question 169: Given Pfizer’s and Moderna’s data from animals showing a wide distribution of LNPs and/or modRNA and given FDA’s participation in the drafting of WHO guidelines (48, 172) suggesting a much narrower distribution, what revisions has FDA proposed or will propose to those document regarding vaccines if any kind that use LNP-technology?............................................................................................... 54 Q169
Question 170: What data has FDA relied upon to validate the assertion that “biodistribution and retention are properties of the LNP rather than the mRNA”?................................................................................................................................................................... 55 Q170
Question 171: Why has the FDA not required data on the biodistribution of the spike protein? That is, have cells been transfected and have subsequently produced the desired protein?....................................................................................................................... 55 Q171
Question 172: Given the novel mechanism of action, delivery and distribution of modRNA vaccines is not contemplated by WHO guidelines (172, 177), please provide a rationale for why they can be used to justify the non-conduct of RNA or protein metabolism or excretion studies on the candidate vaccine formulations?............................................................................................................................................... 56 Q172
Question 173: What studies were conducted to establish that the biodistribution of modRNA incorporated into formulations used in the mRNA 1647 and mRNA 1273 test articles in Moderna’s toxicology and biodistribution studies, is equivalent?........................................ 57 Q173
Question 174: How does the distribution and gene expression of mRNA as lipid-adduct compare with that of non-adducted mRNA?57 Q174
Question 175: Is mRNA as lipid-adduct detected in the multiplex branched DNA (bDNA) assay used to determine levels of mRNA in tissues in Moderna’s biodistribution study?.................................................................................................................................... 57 Q175
Question 176: Does the presence of mRNA as lipid-adduct confound in any way the results and interpretation of Moderna’s biodistribution study and its gene expression?................................................................................................................................................. 57 Q176
Question 177: Given that the formulation of the mRNA 1647 used in Moderna’s toxicology and biodistribution studies appear to differ substantially from mRNA 1273 in ways that likely materially affect LNP physicochemical, distribution and transfection properties, how do studies involving mRNA 1647 support “the approval of SPIKEVAX BLA”?....................................................................................................................... 57 Q177
Question 178: Per Question 170, and in view of the findings in Moderna’s own PK study suggesting construct-dependent kinetics, controverting FDA’s premise of almost exclusively LNP-dependent kinetics, what studies will FDA soliciting to better characterize the PK of the Moderna COVID-19 mRNA vaccine?60 Q178
Question 179: In view of the findings in Moderna’s own PK study suggesting construct-dependent kinetics, controverting FDA’s premise of almost exclusively LNP-dependent kinetics, what guidance will FDA issue regarding the sorts of PK studies needed to support mRNA product approval?60 Q179
Question 180: In view of the findings in Moderna’s own PK study suggesting construct-dependent kinetics, controverting FDA’s premise of almost exclusively LNP-dependent kinetics, what assurance can FDA give about the safety of a product whose approval has relied exclusively on toxicology studies conducted with non-candidate constructs?..................................................................................................................... 60 Q180
Question 181: What in vivo studies did Pfizer or Moderna provide to describe the distribution and kinetics of spike protein production after dosing with COVID-19 modRNA vaccines?...................................................................................................................................... 61 Q181
Question 182: Given reports of vaccinal modRNA or spike protein persistence far longer than indicated by Pfizer’s limited PK data or public health messaging, what animal or human studies have FDA requested of Pfizer and Moderna to better understand the PK of COVID-19 modRNA vaccines and to better inform a risk benefit analysis? Do these include studies using commercially available product?...................................... 61 Q182
Question 183: Given reports of vaccinal modRNA or spike protein persistence far longer than indicated by Pfizer’s limited PK data or public health messaging, what guidance will FDA provide regarding the types of animal and human PK, distribution and expression kinetics studies should be performed for modRNA vaccines or other modRNA gene therapies?.................................................................................................. 61 Q183
Question 184: On what basis did Pfizer “expected” that the components of the vaccine construct are lipids and RNA would not have genotoxic potential? Did FDA challenge this expectation?................................................................................................................... 63 Q184
Question 185: Will FDA correct Dr. Marks’ statement that “Additionally, studies have been conducted in animals using the modified mRNA and lipid nanoparticle together that constitute the vaccine, including the minute quantities of residual DNA fragments left over after DNAse treatment during manufacturing, and demonstrate no evidence for genotoxicity from the vaccine”?.......................................................................... 64 Q185
Question 186: What was the regulatory basis for FDA’s heavy reliance on nonclinical studies involving early modRNA COVID-19 vaccine prototypes, or non-COVID-19 modRNA prototypes, rather than on studies involving test articles of substantially identical composition to the authorized product? To what extent was reliance based on the EUA “totality-of-evidence” standard?...................................................................... 64 Q186
Question 187: How is FDA’s heavy reliance on nonclinical studies involving early modRNA COVID-19 vaccine prototypes, or non-COVID-19 modRNA prototypes, rather than on studies involving test articles of substantially identical composition to the authorized product, compatible with BLA requirements?................................................................................................................................................................... 64 Q187
Question 188: Which nonclinical studies have FDA requested from Pfizer of Moderna to rectify the quality and quantity of the limited studies relied upon under EUA conditions, but would have been insufficient in non-pandemic conditions?................................................ 64 Q188
Question 189: While the dose of ALC-0159 appears to be low compared with the doses associated with genotoxicity (according to the EMA report), what consideration was given to a possible synergistic effect of sub-genotoxic threshold levels of this component with other vaccine components?65 Q189
Question 190: Please confirm the identity of mystery mRNA. To the extent that redaction ever qualified for a (b)(4) exemption, the matter has already been publicly disclosed in the EMA and Moderna documents.................................................................................. 66 Q190
Question 191: Please explain the reason why the genotoxicity study with the “other” mRNA was not described FDA’s document. (151) FDA’s omission does not appear to be a failure by Moderna to report the study to FDA. (152)................................................................ 66 Q191
Question 192: Please provide the study reports for the rat micronucleus assays conducted on the Zika (Study 9800399) and luciferase (Study AF87FU.125012 NGLPICH.BTL) test articles.......................................................................................................................... 67 Q192
Question 193: Please discuss how reductions in reticulocytes and polychromatic cells seen in toxicology and genotoxicity studies are mechanistically related to an increase in micronucleated cells seen in the Zika mRNA genotoxicity study................................................. 67 Q193
Question 194: Do reductions in reticulocytes and polychromatic cells, as well as disturbances in erythropoiesis raise any concerns for bone marrow toxicity? What follow up studies or risk analysis has FDA requested on this topic?........................................................ 67 Q194
Question 195: Given the disclosure (11.4.1) that the sizes of LNPs in a CMV vaccine examined in a biodistribution study were smaller than those found in mRNA-1273, despite misleading statements suggesting that they were the same, what assurances can FDA give that the formulations of the Zika and luciferase mRNA vaccines subjected to genotoxicity tests were identical to mRNA-1273, other than in the modRNA sequence coding for the target antigen/ luciferase?................................................................................................................................................................... 67 Q195
Question 196: Will FDA resolve the discrepancies between the FDA, EMA and Moderna documents regarding which LNP components were tested in these genotoxicity tests......................................................................................................................................... 67 Q196
Question 197: What was the nature of the concern for mutagenic impurities in PEG2000-DMG?....................... 67 Q197
Question 198: Did FDA have a similar concern for mutagenic impurities in PEG2000-DMG as did EMA?........... 68 Q198
Question 199: How was this concern for mutagenic impurities in PEG2000-DMG resolved?............................. 68 Q199
Question 200: Given that the evaluation of the potential presence of mutagenic impurities in PEG2000-DMG was to be provided post-approval, when exactly did this occur?................................................................................................................................................... 68 Q200
Question 201: If mutagenic impurities did exist in PEG2000-DMG prior to resolution of this issue, how many doses of mRNA-1273 (and to how many people) had been administered either in clinical trials or post approval/ authorization?.................................................. 68 Q201
Question 202: Has any risk assessment been conducted or requested by FDA to assess whether synergistic effects occurred between any subthreshold mutagenicity of impurities in PEG2000-DMG and any effects from other vaccine components or impurities?...... 68 Q202
Question 203: What was the nature of the concern for benzene or mutagenic impurities in SM-102?................. 68 Q203
Question 204: Did FDA have a similar concern for benzene or mutagenic impurities in SM-102 as did EMA?..... 68 Q204
Question 205: How was this concern for benzene or mutagenic impurities in SM-102 resolved?........................ 68 Q205
Question 206: Given that the risk assessment for the presence of benzene in SM-102 was to be provided by June 30 2021, when exactly did this occur?68 Q206
Question 207: If benzene or mutagenic impurities did exist in SM-102 prior to resolution of this issue, how many doses (and to how many people) of mRNA-1273 had been administered either in clinical trials or post approval/ authorization?.......................................... 68 Q207
Question 208: Has any risk assessment been conducted or requested by FDA to assess whether synergistic effects occurred between any benzene and subthreshold mutagenicity of impurities in SM-102 and any effects from other vaccine components or impurities?68 Q208
Question 209: Will FDA release the full original reports for all Moderna and Pfizer toxicological studies supporting the various EUA or BLA’s for their COVID-19 mRNA vaccines?......................................................................................................................................... 68 Q209
Question 210: What is the identity of the ingredient “PG” used in some of Moderna’s repeat-dose toxicology studies and in its biodistribution study? Is this polyethylene glycol (PEG)?........................................................................................................................... 68 Q210
Question 211: Given the disclosure (11.4.1) that the sizes of LNPs in a CMV vaccine examined in a biodistribution study were smaller than those found in mRNA-1273, despite misleading statements suggesting that they were the same, what assurances can FDA give that the formulations of the Zika and luciferase mRNA vaccines subjected to genotoxicity tests were identical to mRNA-1273, other than in the modRNA sequence coding for the target antigen/ luciferase?................................................................................................................................................................... 68 Q211
Question 212: Was FDA aware of the procedural/methodological limitations in Moderna’s only repeat-dose toxicology study on its SARS-CovV-2 mRNA candidate described in the EMA document?................................................................................................... 69 Q212
Question 213: The procedural/methodological limitations indicated by EMA for Moderna’s only repeat-dose toxicology study on its SARS-CovV-2 mRNA candidate appear to extend beyond non-compliance with GLP. What was the nature of these limitations?......... 69 Q213
Question 214: The absence in FDA’s document of a qualifying statement similar to that in the EMA document appears to be a material omission possibly affecting the interpretation of the body of nonclinical data. Please justify or comment....................................... 69 Q214
Question 215: What standard of evidence did FDA ascribe to the comparison of data from a procedurally, methodologically, and inadequate study of Moderna’s SARS-Cov-2 vaccine candidate to data from studies on other mRNA products?............................................... 69 Q215
Question 216: Even if, per Question 215, the quality of data from the only repeat-dose toxicology study of Moderna’s COVID-19 vaccine candidate was sufficient to meet the EUA “totality of evidence” standard, did FDA consider that this study met BLA requirements?69 Q216
Question 217: Were these large unstained cells in Pfizer’s repeat dose toxicity study reported to FDA?Did FDA seek clarification as to their nature? What was Pfizer’s response?........................................................................................................................................ 69 Q217
Question 218: Referencing Question 61 (section 3.6), what other differences in the results from nonclinical studies were found between the V8 and V9 Pfizer product versions?......................................................................................................................................... 69 Q218
Question 219: Did FDA challenge Pfizer’s assertion based on WHO 2005 guideline, that genotoxicity would not be needed?71 Q219
Question 220: Will FDA work to remove ambiguities in their own guidelines related to the conduct of genotoxicity and carcinogenicity studies for modRNA pro-vaccines?.................................................................................................................................................... 71 Q220
Question 221: Although FDA did not identify any cancer signals using Empirical Bayesian Data Mining, (EBDM) their analysis was recently suggested to be seriously flawed. (204) Please provide full details of all signals generated by EBDM, including where the threshold is set to EB05>1, and the RGPS procedure within the Empirica software is used to adjust for masking, using the ER05>1 criteria?..................................... 72 Q221
Question 222: Did Pfizer or Moderna identify within their modRNA sequences any unexpected ORFs, including frameshift sequences per the WHO guidelines (71)? What were these sequences and when was this information provided?................................................... 73 Q222
Question 223: Given the submission and publication of the Mulroney paper in January and December 2023 respectively, when did FDA first learn about the findings in this paper and from whom?........................................................................................................... 74 Q223
Question 224: Given the production of neo-antigens or unwanted immune responses that “may require a redesign of the mRNA sequence” according to this WHO committee (214) and the description of the modRNA vaccines as “error-prone” in the Mulroney press release, (210) does FDA consider the non-selective N1-methylpseudouridylation of the Moderna and Pfizer COVID-19 vaccines to be an inherent design flaw?....... 74 Q224
Question 225: What discussions have taken place between Pfizer or Moderna and FDA or other government agencies or entities regarding the need to redesign the COVID-19 vaccines as well as other vaccines employing the same technology?........................... 74 Q225
Question 226: What will be the regulatory pathway for the introduction of redesigned modRNA-based vaccines?74 Q226
Question 227: Is FDA attempting to characterize the frameshift proteins in terms of their primary, secondary and tertiary structures, their glycosylation patterns, the sites and kinetics of their production, their pharmaco- and toxico-kinetics?................................................. 74 Q227
Question 228: What efforts have been made or are are underway to study the identity, pharmacology and toxicology of these frameshift proteins?74 Q228
Question 229: What studies have been conducted and which in silico tools been utilized to screen for likely interactions of these frameshift proteins in the body?.......................................................................................................................................................... 74 Q229
Question 230: In addition to in vitro and animal models, are these studies being conducted in humans? How do the actions of the frameshift proteins vary by age, gender, genetic make-up and comorbidity?............................................................................................ 74 Q230
Question 231: Given their possible chimeric nature, what efforts are underway to determine whether there are synergistic pharmacologic, immunologic or toxicologic effects between the frameshift proteins and the intended in-frame spike proteins?........................... 74 Q231
Question 232: Have genomic and proteomic databases and tools such as BLAST been interrogated to determine if there are any homologies between the proposed frameshift proteins and peptides and known proteins?..................................................................... 74 Q232
Question 233: What efforts are underway to determine if there are associations between the formation and type of frameshift proteins and adverse events that have been already been experienced or reported?......................................................................................... 74 Q233
Question 234: What efforts are underway to determine if there are likely to be long-term consequences of these frameshift proteins?74 Q234
Question 235: What efforts are underway to monitor for the occurrence of long-term consequences of these frameshift proteins?74 Q235
Question 236: What efforts are underway to determine methods for the diagnosis of possible harms caused by frameshift-proteins and treatments to limit, prevent or treat harm?.................................................................................................................................. 75 Q236
Question 237: Did FDA express the same concern as EMA regarding vaccine-elicited autoimmune responses? Did Pfizer or Moderna submit a discussion on this topic to FDA, per the invitation extended to Pfizer in the EMA document? When? Please supply a copy...... 75 Q237
Question 238: In view of the evidence for off-target immune responses elicited by frameshift proteins described by Mulroney et al., as well as the concerns expressed in WHO and EMA documents, have Pfizer or Moderna been asked to submit, or have Pfizer or Moderna already submitted any risk assessments related to the production of frameshift proteins? When? Please supply a copy................................................. 75 Q238
Question 239: Is FDA aware of, or has it solicited or received from Pfizer, Moderna or other research entity, a full characterization of the off-target immune response elicited by frameshift proteins, alone or in combination with on-target proteins? In addition to characterization of the cellular response, as was partly provided in the Mulroney paper, does this work also include a characterization of the humoral response, which was not described by Mulroney et al.75 Q239
Question 240: What actions will FDA be taking to correct these potentially misleading statements by including appropriate labelling language describing the production of uncharacterized off-target frameshift proteins with unknown toxicology capable of eliciting uncharacterized off-target immune response of yet unknown clinical significance?....................................................................................................................... 75 Q240
Question 241: What other steps will FDA take to inform the medical community and the lay public of he potential risks associated with the production of frameshift proteins?...................................................................................................................................... 76 Q241
Question 242: What analysis has FDA conducted, or will conduct to investigate the root causes and systems failures for their apparent failure and/or that of the manufacturers to identify, detect, report, and investigate the formation of frameshift proteins and their potential risks?76 Q242
Question 243: If appropriate, what corrective actions has or will FDA implement within its own organization to ensure that this failure will not be repeated?76 Q243
Question 244: If appropriate, what regulatory actions has or will FDA implement regarding the manufacturers of modRNA COVID-19 vaccines to ensure that this failure will not be repeated?.................................................................................................................... 76 Q244
Question 245: Have Pfizer or Moderna submitted any data or risk analysis concerning the possible formation of other kinds of cryptic proteins (cryptides or crypteins) such as those produced from alternate start sites(67, 211) or proteolytic cleavage.(212).................... 76 Q245
Question 246: Along the lines of the questions enumerated above, will FDA conduct full assessments of past or future harms associated with these proteins, identity root causes for the failure to identify this problem sooner, identify corrective actions to prevent future failures, and to inform the public of these findings?................................................................................................................................................................... 76 Q246
Question 247: Will FDA conduct or support the development of methods for the diagnosis, prevention and treatment of harm related to frameshift or other possible cryptic proteins?.............................................................................................................................. 76 Q247
Question 248: Will FDA acknowledge the biological reality of these products and regulate them as gene therapies?77 Q248
19. Revision history
V1 091325
20. References
1. Ladapo JA. Letter to Robert M. Califf, MD, MACC Commissioner U.S. Food and Drug Administration and Mandy Cohen, MD, MPH, Director Centers for Disease Control and Prevention. 2023 Dec 6. at https://www.floridahealth.gov/about/_documents/12-06-2023-DOH-Letter-to-FDA-RFI-on-COVID-19-Vaccines.pdf .)
2. McKernan K, Helbert, Y., Kane, L. T., McLaughlin, S. Sequencing of bivalent Moderna and Pfizer mRNA vaccines reveals nanogram to microgram quantities of expression vector dsDNA per dose. . OSF Preprints 2023. Epub Sep 25 http://doi.org/10.31219/osf.io/b9t7m
3. Speicher DJ, Rose J, Gutschi LM, Wiseman DM, McKernan K. DNA fragments detected in monovalent and bivalent Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada: Exploratory dose response relationship with serious adverse events. OSF Preprints 2023. Epub Oct 19 http://doi.org/10.31219/osf.io/mjc97
4. Marks P. Letter to Florida Surgeon General Ladapo. 2023. at https://www.fda.gov/media/174875/download .)
5. Florida Health. Florida State Surgeon General Calls for Halt in the Use of COVID-19 mRNA Vaccines | Florida Department of Health. 2024 Jan 3. at https://www.floridahealth.gov/newsroom/2024/01/20240103-halt-use-covid19-mrna-vaccines.pr.html .)
6. König B, Kirchner JO. Methodological Considerations Regarding the Quantification of DNA Impurities in the COVID-19 mRNA Vaccine Comirnaty®. Methods and Protocols 2024; 7. Epub http://doi.org/10.3390/mps7030041
7. König BK, J. O. Communication on Methodological Considerations Regarding the Quantification of DNA Impurities in the COVID-19 mRNA Vaccine Comirnaty®. Methods Protoc. 2024, 7, 41. Preprints 2024:2024111912. Epub Nov 29 http://doi.org/https://doi.org/10.20944/preprints202411.1912.v2
https://doi.org/10.20944/preprints202411.1912.v5
8. Kämmerer U SV, Steger K. BioNTech RNA-Based COVID-19 Injections Contain Large Amounts Of Residual DNA Including An SV40 Promoter/Enhancer Sequence. Science, Public Health Policy and the Law 2024; 5:2019-24. Epub Dec 3 , https://publichealthpolicyjournal.com/biontech-rna-based-covid-19-injections-contain-large-amounts-of-residual-dna-including-an-sv40-promoter-enhancer-sequence/
9. Raoult D. Confirmation of the presence of vaccine DNA in the Pfizer anti-COVID-19 vaccine. HAL Open Science 2024. Epub , https://hal.science/hal-04778576
10. Buckhaults P. Dr. Phillip Buckhaults. Testimony to South Carolina Senate Medical Affairs Ad-Hoc Committee on DHEC. 2023 Sept 13. at https://www.youtube.com/watch?v=IEWHhrHiiTY
https://video.scstatehouse.gov/mp4/20230912SMedicalAffairsSenateCommittee13489_1.mp4
)
11. Wang TJ, Kim A, Kim K. A rapid detection method of replication-competent plasmid DNA from COVID-19 mRNA vaccines for quality control. Journal of High School Science 2024; 8:427-39. Epub , https://jhss.scholasticahq.com/article/127890-a-rapid-detection-method-of-replication-competent-plasmid-dna-from-covid-19-mrna-vaccines-for-quality-control
12. Wu KM. A New Classification of Prodrugs: Regulatory Perspectives. Pharmaceuticals (Basel) 2009; 2:77-81. Epub 20091014 http://doi.org/10.3390/ph2030077
13. Cosentino M, Marino F. Understanding the Pharmacology of COVID-19 mRNA Vaccines: Playing Dice with the Spike? Int J Mol Sci 2022; 23:10881. Epub Sept 17 http://doi.org/10.3390/ijms231810881
14. Banoun H. mRNA: Vaccine or Gene Therapy? The Safety Regulatory Issues. Int J Mol Sci 2023; 24. Epub http://doi.org/10.3390/ijms241310514
15. FDA. COMIRNATY Package Insert 2023-2024 Formula. 2023 Sept 11. at https://www.fda.gov/media/151707/download?attachment .)
16. FDA. COMIRNATY Package Insert 2024-2025 Formula. 2024 August 22. at https://www.fda.gov/media/151707/download?attachment .)
17. FDA. Spikevax Package Insert 2023-2024 Formula. 2023 Sep 11. at https://www.fda.gov/media/155675/download?attachment .)
18. FDA. Spikevax Patient Package Insert 2024-2025 Formula. 2024 August 22. at https://www.fda.gov/media/155762/download?attachment .)
19. FDA. Guidance for Industry Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products. 1998 May 15. at https://www.fda.gov/media/71655/download .)
20. FDA. Demonstrating Substantial Evidence of Effectiveness for Human Drug and Biological Products Guidance for Industry DRAFT GUIDANCE 2019 Dec. at https://www.fda.gov/media/133660/download .)
21. FDA. Demonstrating Substantial Evidence of Effectiveness With One Adequate and Well-Controlled Clinical Investigation and Confirmatory Evidence Guidance for Industry DRAFT GUIDANCE. 2023 Sept 19. at https://www.fda.gov/media/172166/download .)
22. FDA. Communications From Firms to Health Care Providers Regarding Scientific Information on Unapproved Uses of Approved/Cleared Medical Products Questions and Answers Guidance for Industry DRAFT GUIDANCE. 2023 Oct. at https://www.fda.gov/media/173172/download .)
23. FDA. FACT SHEET FOR HEALTHCARE PROVIDERS ADMINISTERING VACCINE (VACCINATION PROVIDERS) EMERGENCY USE AUTHORIZATION (EUA) OF THE PFIZER-BIONTECH COVID-19 VACCINE TO PREVENT CORONAVIRUS DISEASE 2019 (COVID-19) FOR 5 THROUGH 11 YEARS OF AGE DILUTE BEFORE USE. 2022 Jan 3. (Accessed Mar 11, 2022, at https://www.fda.gov/media/153714/download .)
24. Moderna. FACT SHEET FOR HEALTHCARE PROVIDERS ADMINISTERING VACCINE (VACCINATION PROVIDERS) EMERGENCY USE AUTHORIZATION (EUA) OF THE MODERNA COVID-19 VACCINE TO PREVENT CORONAVIRUS DISEASE 2019 (COVID-19). 2021. Epub , https://www.fda.gov/media/144637/download
25. CDC. Vaccination Considerations for People Pregnant or Breastfeeding. 2023 Nov 3. at https://www.cdc.gov/coronavirus/2019-ncov/vaccines/recommendations/pregnancy.html .)
26. FDA. Facts about COVID-19. 2023 Oct 20. at https://www.fda.gov/news-events/rumor-control/facts-about-covid-19 .)
27. CDC. Clinical Guidance for COVID-19 Vaccination: Considerations involving pregnancy, lactation, and fertility. 2023 Nov 3. at https://www.cdc.gov/vaccines/covid-19/clinical-considerations/interim-considerations-us.html#pregnancy-fertility .)
28. CDC. COVID-19 Vaccination for Women Who Are Pregnant or Breastfeeding. 2024 Sept 10. at https://www.cdc.gov/covid/vaccines/pregnant-or-breastfeeding.html?CDC_AAref_Val=https://www.cdc.gov/coronavirus/2019-ncov/vaccines/recommendations/pregnancy.html .)
29. Polack FP, Thomas SJ, Kitchin N, et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med 2020; 383:2603-15. Epub 2020/12/11 http://doi.org/10.1056/NEJMoa2034577
30. EMA. Assessment report: Comirnaty - Pfizer. 2021 Feb 19. at https://www.ema.europa.eu/en/documents/assessment-report/comirnaty-epar-public-assessment-report_en.pdf .)
31. FDA. Guidance for Industry. Q5E Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process. 2005 June. at https://www.fda.gov/media/71489/download .)
32. FDA. Food and Drug Administration. Development and Licensure of Vaccines to Prevent COVID-19: Guidance for Industry. 2020 June 30. (Accessed 2021 Jan 31, at https://www.fda.gov/media/139638/download .)
33. FDA. Food and Drug Administration. Emergency Use Authorization for Vaccines to Prevent COVID-19 Guidance for Industry 2020 October 6. (Accessed 2023 July 5, at https://web.archive.org/web/20201007024021/https://www.fda.gov/media/142749/download .)
34. FDA. Emergency Use Authorization of Medical Products and Related Authorities. 2017. 2022, at https://www.fda.gov/media/97321/download .)
35. Weir J. Licensure and Emergency Use Authorization of Vaccines to Prevent COVID-19: Chemistry, Manufacturing,and Controls (CMC) Considerations. Vaccines and Related Biological Products Advisory Committee (10/22/2020). 2020 Oct 22. at https://www.fda.gov/media/143353/download .)
36. FDA. Transcript: 161st Vaccines and Related Biological Products Advisory Committee (VRBPAC) Meeting. 2020 Oct 22. at https://www.fda.gov/media/143982/download .)
37. Guetzkow J, Levi, R. Effect of mRNA Vaccine Manufacturing Processes on Efficacy and Safety Still an Open Question
Rapid response to:: Covid-19: Researchers face wait for patient level data from Pfizer and Moderna vaccine trials. BMJ 2023. Epub May 13 , https://www.bmj.com/content/378/bmj.o1731/rr-2
https://www.bmj.com/content/378/bmj.o1731/rapid-responses
38. FDA. Briefing Document: Vaccines and Related Biological Products Advisory Committee Meeting October 22, 2020. Development, authorization and licensure of vaccines to prevent COVID-19. 2020 Oct 22. (Accessed July 19, 2021, at https://www.fda.gov/media/142723/download .)
39. Pfizer. BNT162b2 (PF-07302048) Comparability Report for PPQ Drug Product Lots. INX100451158. Post approval commitments by sponsor for Comirnaty in relation to batch analysis for drug product batches manufactured at Pfizer, released by TGA FOI 3659 June 3 2022. 2021. at https://www.tga.gov.au/sites/default/files/2022-08/foi-3659-04.pdf .)
40. MHRA. Medicines and Healthcare products Regulatory Agency. Internal review of FOI 23/510: Response to Mr. NH Hunt. 2023 Sep 21.
41. FDA. Considerations for the Use of Real-World Data and Real-World Evidence to Support Regulatory Decision-Making for Drug and Biological Products Guidance for Industry. 2023 Aug 30. at https://www.fda.gov/media/171667/download .)
42. Patel HK, Zhang K, Utegg R, et al. Characterization of BNT162b2 mRNA to Evaluate Risk of Off-Target Antigen Translation. J Pharm Sci 2023. Epub 20230112 http://doi.org/10.1016/j.xphs.2023.01.007
43. EMA. CHMP Assessment Report for the Post-Authorisation Measure REC 027, Comirnaty (EMA/CHMP/284816/2021). Released per ASK-148075, October 25, 2023. 2021 May 20.
44. EMA. Cyberattack on the European Medicines Agency. 2020 Dec 9. at https://www.ema.europa.eu/en/news/cyberattack-european-medicines-agency .)
45. Tinari S. The EMA covid-19 data leak, and what it tells us about mRNA instability. BMJ 2021; 372:n627. Epub 2021/03/12 http://doi.org/10.1136/bmj.n627
46. EMA. CHMP Type IB variation report, Comirnaty EMA/CHMP/50784/2022. Released per ASK-148075, October 25, 2023. 2022 March 31.
47. FDA. Summary Basis for Regulatory Action COMIRNATY. 2021 Nov 8. at https://www.fda.gov/media/151733/download .)
48. WHO. Guidelines on the quality, safety and efficacy of plasmid DNA vaccines. Annex 2. TRS No 1028 Replacement of Annex 1 of WHO Technical Report Series, No. 941. 2021 Mar 10. at https://cdn.who.int/media/docs/default-source/biologicals/vaccine-standardization/dna-vaccines/annex-2_dna_who_trs_1028_web-(1).pdf
https://www.who.int/publications/m/item/plasmid-dna-vaccines-annex-2-trs-no-1028 .)
49. WHO. WHO Technical Report Series No 941, 2007. Annex 1. Guidelines for assuring the quality and nonclinical safety evaluation of DNA vaccines. 2007. at https://cdn.who.int/media/docs/default-source/biologicals/vaccine-quality/guidelines-for-assuring-the-quality-and-non-clinical-safety-evaluation-of-dna-vaccines70ee1b3e-88a6-40af-8989-fbff8304a377.pdf?sfvrsn=521ee591_1&download=true .)
50. WHO. Annex 3. Recommendations for the evaluation of animal cell cultures as substrates for the manufacture of biological medicinal products and for the characterization of cell banks Replacement of Annex 1 of WHO Technical Report Series, No. 878. 2013. at https://cdn.who.int/media/docs/default-source/biologicals/documents/trs_978_annex_3.pdf?sfvrsn=fe61af77_3&download=true .)
51. Kurth R. Risk potential of the chromosomal insertion of foreign DNA. Ann N Y Acad Sci 1995; 772:140-51. Epub http://doi.org/10.1111/j.1749-6632.1995.tb44739.x
52. Temin HM. Overview of biological effects of addition of DNA molecules to cells. J Med Virol 1990; 31:13-7. Epub http://doi.org/10.1002/jmv.1890310105
53. Petricciani JC, Regan PJ. Risk of neoplastic transformation from cellular DNA: calculations using the oncogene model. Dev Biol Stand 1987; 68:43-9. Epub ,
54. Coffin JM. Molecular mechanisms of nucleic acid integration. J Med Virol 1990; 31:43-9. Epub http://doi.org/10.1002/jmv.1890310109
55. Nichols WW, Ledwith BJ, Manam SV, Troilo PJ. Potential DNA vaccine integration into host cell genome. Ann N Y Acad Sci 1995; 772:30-9. Epub 1995/11/27 http://doi.org/10.1111/j.1749-6632.1995.tb44729.x
56. Petricciani JC, Horaud FN. DNA, dragons and sanity. Biologicals 1995; 23:233-8. Epub http://doi.org/10.1006/biol.1995.0039
57. Cooper G, Sunderland, MA. The Nuclear Envelope and Traffic between the Nucleus and Cytoplasm. The Cell: A Molecular Approach. 2nd ed: Sinauer Associates; 2000.
58. Cooper G, Sunderland, MA. The Nucleus during Mitosis. The Cell: A Molecular Approach. 2nd ed: Sinauer Associates; 2000.
59. Boettcher B, Barral Y. The cell biology of open and closed mitosis. Nucleus 2013; 4:160-5. Epub 20130415 http://doi.org/10.4161/nucl.24676
60. Faurez F, Dory D, Le Moigne V, Gravier R, Jestin A. Biosafety of DNA vaccines: New generation of DNA vectors and current knowledge on the fate of plasmids after injection. Vaccine 2010; 28:3888-95. Epub 20100404 http://doi.org/10.1016/j.vaccine.2010.03.040
61. Lechardeur D, Lukacs GL. Nucleocytoplasmic transport of plasmid DNA: a perilous journey from the cytoplasm to the nucleus. Hum Gene Ther 2006; 17:882-9. Epub http://doi.org/10.1089/hum.2006.17.882
62. Lim S, Yocum RR, Silver PA, Way JC. High spontaneous integration rates of end-modified linear DNAs upon mammalian cell transfection. Sci Rep 2023; 13:6835. Epub 20230426 http://doi.org/10.1038/s41598-023-33862-0
63. FDA. Guidance for Industry. Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications 2010 February. at https://www.fda.gov/media/78428/download .)
64. FDA. Food and Drug Administration. Considerations for Plasmid DNA Vaccines for Infectious Disease Indications. Guidance for Industry. 2007 November. (Accessed Jul 29, 2021, at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-plasmid-dna-vaccines-infectious-disease-indications
https://www.fda.gov/media/73667/download .)
65. Sheng-Fowler L, Lewis AM, Jr., Peden K. Issues associated with residual cell-substrate DNA in viral vaccines. Biologicals 2009; 37:190-5. Epub 20090314 http://doi.org/10.1016/j.biologicals.2009.02.015
66. Sahin U, Kariko K, Tureci O. mRNA-based therapeutics--developing a new class of drugs. Nat Rev Drug Discov 2014; 13:759-80. Epub 2014/09/23 http://doi.org/10.1038/nrd4278
67. Reichmuth AM, Oberli MA, Jaklenec A, Langer R, Blankschtein D. mRNA vaccine delivery using lipid nanoparticles. Ther Deliv 2016; 7:319-34. Epub 2016/04/15 http://doi.org/10.4155/tde-2016-0006
68. Dalby B, Cates S, Harris A, et al. Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods 2004; 33:95-103. Epub http://doi.org/10.1016/j.ymeth.2003.11.023
69. Dean DA, Dean BS, Muller S, Smith LC. Sequence requirements for plasmid nuclear import. Exp Cell Res 1999; 253:713-22. Epub http://doi.org/10.1006/excr.1999.4716
70. Vacik J, Dean BS, Zimmer WE, Dean DA. Cell-specific nuclear import of plasmid DNA. Gene Ther 1999; 6:1006-14. Epub http://doi.org/10.1038/sj.gt.3300924
71. WHO. Evaluation of the quality, safety and efficacy of messenger RNA vaccines for the prevention of infectious diseases: regulatory considerations. WHO/BS/2021.2402. 2021. (Accessed June 16, 2022, at https://cdn.who.int/media/docs/default-source/biologicals/call-for-comments/bs.2021.bs2402_who-regulatory-considerations-for-mrna-vaccines_final.pdf .)
72. Fertig TE, Chitoiu L, Marta DS, et al. Vaccine mRNA Can Be Detected in Blood at 15 Days Post-Vaccination. Biomedicines 2022; 10. Epub http://doi.org/10.3390/biomedicines10071538
73. Castruita JAS, Schneider UV, Mollerup S, et al. SARS-CoV-2 spike mRNA vaccine sequences circulate in blood up to 28 days after COVID-19 vaccination. Apmis 2023; 131:128-32. Epub 20230129 http://doi.org/10.1111/apm.13294
74. Bansal S, Perincheri S, Fleming T, et al. Cutting Edge: Circulating Exosomes with COVID Spike Protein Are Induced by BNT162b2 (Pfizer-BioNTech) Vaccination prior to Development of Antibodies: A Novel Mechanism for Immune Activation by mRNA Vaccines. J Immunol 2021; 207:2405-10. Epub 2021/10/17 http://doi.org/10.4049/jimmunol.2100637
75. Röltgen K, Nielsen SCA, Silva O, et al. Immune imprinting, breadth of variant recognition and germinal center response in human SARS-CoV-2 infection and vaccination. Cell 2022 Jan 24. Epub http://doi.org/10.1016/j.cell.2022.01.018
76. Brogna C, Cristoni S, Marino G, et al. Detection of recombinant Spike protein in the blood of individuals vaccinated against SARS-CoV-2: Possible molecular mechanisms. PROTEOMICS – Clinical Applications 2023; n/a:2300048. Epub http://doi.org/https://doi.org/10.1002/prca.202300048
77. Ota N, Itani M, Aoki T, et al. Expression of SARS-CoV-2 spike protein in cerebral Arteries: Implications for hemorrhagic stroke Post-mRNA vaccination. Journal of Clinical Neuroscience 2025; 136:111223. Epub http://doi.org/https://doi.org/10.1016/j.jocn.2025.111223
78. Pateev I, Seregina K, Ivanov R, Reshetnikov V. Biodistribution of RNA Vaccines and of Their Products: Evidence from Human and Animal Studies. Biomedicines 2023; 12. Epub 20231226 http://doi.org/10.3390/biomedicines12010059
79. Sattar S, Kabat J, Jerome K, et al. Nuclear translocation of spike mRNA and protein is a novel pathogenic feature of SARS-CoV-2. bioRxiv 2022:2022.09.27.509633. Epub Sep 27 http://doi.org/10.1101/2022.09.27.509633
80. Domazet-Lošo T. mRNA vaccines: Why is the biology of retroposition ignored? ResearchGate 2021. Epub Jul 24 http://doi.org/10.31219/osf.io/uwx32
81. Nicolau M, Picault N, Moissiard G. The Evolutionary Volte-Face of Transposable Elements: From Harmful Jumping Genes to Major Drivers of Genetic Innovation. Cells 2021; 10. Epub 20211029 http://doi.org/10.3390/cells10112952
82. Lafon-Hughes L. Towards Understanding Long COVID: SARS-CoV-2 Strikes the Host Cell Nucleus. Pathogens 2023; 12. Epub 20230606 http://doi.org/10.3390/pathogens12060806
83. Zhang L, Richards A, Barrasa MI, et al. Reverse-transcribed SARS-CoV-2 RNA can integrate into the genome of cultured human cells and can be expressed in patient-derived tissues. Proceedings of the National Academy of Sciences 2021; 118:e2105968118. Epub http://doi.org/10.1073/pnas.2105968118
84. Zhang L, Bisht P, Flamier A, et al. LINE1-Mediated Reverse Transcription and Genomic Integration of SARS-CoV-2 mRNA Detected in Virus-Infected but Not in Viral mRNA-Transfected Cells. Viruses 2023; 15. Epub 20230225 http://doi.org/10.3390/v15030629
85. Sarkar AA. Eminent MIT Scientists Defend Controversial SARS-CoV-2 Genome Integration Results. GEN - Genetic Engineering and Biotechnology News 2021. Epub May 13 http://doi.org/www.genengnews.com/insights/eminent-mit-scientists-defend-controversial-sars-cov-2-genome-integration-results
86. Aldén M, Olofsson Falla F, Yang D, et al. Intracellular Reverse Transcription of Pfizer BioNTech COVID-19 mRNA Vaccine BNT162b2 In Vitro in Human Liver Cell Line. Current Issues in Molecular Biology 2022; 44. Epub http://doi.org/10.3390/cimb44030073
87. Baldwin ET, Gotte M, Tchesnokov EP, et al. Human endogenous retrovirus-K (HERV-K) reverse transcriptase (RT) structure and biochemistry reveals remarkable similarities to HIV-1 RT and opportunities for HERV-K-specific inhibition. Proc Natl Acad Sci U S A 2022; 119:e2200260119. Epub 20220630 http://doi.org/10.1073/pnas.2200260119
88. Liu H, Bergant V, Frishman G, et al. Influenza A Virus Infection Reactivates Human Endogenous Retroviruses Associated with Modulation of Antiviral Immunity. Viruses 2022; 14. Epub 20220721 http://doi.org/10.3390/v14071591
89. Petrone V, Fanelli M, Giudice M, et al. Expression profile of HERVs and inflammatory mediators detected in nasal mucosa as a predictive biomarker of COVID-19 severity. Front Microbiol 2023; 14:1155624. Epub 20230522 http://doi.org/10.3389/fmicb.2023.1155624
90. Charvet B, Brunel J, Pierquin J, et al. SARS-CoV-2 awakens ancient retroviral genes and the expression of proinflammatory HERV-W envelope protein in COVID-19 patients. iScience 2023; 26:106604. Epub 20230407 http://doi.org/10.1016/j.isci.2023.106604
91. Di J, Du Z, Wu K, et al. Biodistribution and Non-linear Gene Expression of mRNA LNPs Affected by Delivery Route and Particle Size. Pharm Res 2022; 39:105-14. Epub 20220126 http://doi.org/10.1007/s11095-022-03166-5
92. Phua KK, Leong KW, Nair SK. Transfection efficiency and transgene expression kinetics of mRNA delivered in naked and nanoparticle format. J Control Release 2013; 166:227-33. Epub 20130107 http://doi.org/10.1016/j.jconrel.2012.12.029
93. Turnbull IC, Eltoukhy AA, Fish KM, et al. Myocardial Delivery of Lipidoid Nanoparticle Carrying modRNA Induces Rapid and Transient Expression. Mol Ther 2016; 24:66-75. Epub 20151016 http://doi.org/10.1038/mt.2015.193
94. Ledwith BJ, Manam S, Troilo PJ, et al. Plasmid DNA vaccines: investigation of integration into host cellular DNA following intramuscular injection in mice. Intervirology 2000; 43:258-72. Epub http://doi.org/10.1159/000053993
95. Ledwith BJ, Manam S, Troilo PJ, et al. Plasmid DNA vaccines: assay for integration into host genomic DNA. Dev Biol (Basel) 2000; 104:33-43. Epub ,
96. Sheng-Fowler L, Cai F, Fu H, et al. Tumors induced in mice by direct inoculation of plasmid DNA expressing both activated H-ras and c-myc. Int J Biol Sci 2010; 6:151-62. Epub 20100329 http://doi.org/10.7150/ijbs.6.151
97. Sheng-Fowler L, Tu W, Fu H, et al. A mouse strain defective in both T cells and NK cells has enhanced sensitivity to tumor induction by plasmid DNA expressing both activated H-Ras and c-Myc. PLoS One 2014; 9:e108926. Epub 20141010 http://doi.org/10.1371/journal.pone.0108926
98. Sheng-Fowler L, Tu W, Phy K, et al. Evaluating the sensitivity of newborn rats and newborn hamsters to oncogenic DNA. Biologicals 2023; 84:101724. Epub 20231116 http://doi.org/10.1016/j.biologicals.2023.101724
99. Van Craenenbroeck K, Vanhoenacker P, Haegeman G. Episomal vectors for gene expression in mammalian cells. Eur J Biochem 2000; 267:5665-78. Epub http://doi.org/10.1046/j.1432-1327.2000.01645.x
100. Spadafora C. Sperm-Mediated Transgenerational Inheritance. Front Microbiol 2017; 8:2401. Epub 20171204 http://doi.org/10.3389/fmicb.2017.02401
101. Wang XY, Zhang JH, Zhang X, et al. Impact of different promoters on episomal vectors harbouring characteristic motifs of matrix attachment regions. Sci Rep 2016; 6:26446. Epub 20160526 http://doi.org/10.1038/srep26446
102. Ishii KJ, Kawagoe T, Koyama S, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 2008; 451:725-9. Epub http://doi.org/10.1038/nature06537
103. Lee G-H, Lim S-G. CpG-Adjuvanted Hepatitis B Vaccine (HEPLISAV-B®) Update. Expert Rev Vaccines 2021; 20:487-95. Epub http://doi.org/10.1080/14760584.2021.1908133
104. Martin JE, Sullivan NJ, Enama ME, et al. A DNA vaccine for Ebola virus is safe and immunogenic in a phase I clinical trial. Clin Vaccine Immunol 2006; 13:1267-77. Epub 20060920 http://doi.org/10.1128/CVI.00162-06
105. Gaitzsch E, Czermak T, Ribeiro A, et al. Double-stranded DNA induces a prothrombotic phenotype in the vascular endothelium. Sci Rep 2017; 7:1112. Epub 20170425 http://doi.org/10.1038/s41598-017-01148-x
106. Klein N. COVID-19 Vaccine Safety Surveillance: Summary from VSD RCA. ACIP September 12 2023. 2023 September 12. at https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2023-09-12/07-COVID-Klein-508.pdf .)
107. Heil M. Self-DNA driven inflammation in COVID-19 and after mRNA-based vaccination: lessons for non-COVID-19 pathologies. Front Immunol 2024; 14:1259879. Epub 20240219 http://doi.org/10.3389/fimmu.2023.1259879
108. Ma X, Xin D, She R, et al. Novel insight into cGAS-STING pathway in ischemic stroke: from pre- to post-disease. Front Immunol 2023; 14:1275408. Epub Oct 17 http://doi.org/10.3389/fimmu.2023.1275408
109. Lu Y, Matuska K, Nadimpalli G, et al. Evaluation of Stroke Risk Following COVID-19 mRNA Bivalent Vaccines Among U.S. Adults Aged ≥65 Years. medRxiv 2023:2023.10.10.23296624. Epub Oct 15 http://doi.org/10.1101/2023.10.10.23296624
110. Xu S, Sy LS, Hong V, et al. Ischemic Stroke after Bivalent COVID-19 Vaccination: A Self-Controlled Case Series Study. medRxiv 2023:2023.10.12.23296968. Epub Oct 15 http://doi.org/10.1101/2023.10.12.23296968
111. Finol E, Krul SE, Hoehn SJ, Crespo-Hernández CE. The mRNACalc web server accounts for the hypochromicity of modified nucleosides and enables the accurate quantification of nucleoside-modified mRNA. bioRxiv 2023:2023.07.27.550903. Epub http://doi.org/10.1101/2023.07.27.550903
112. USP. Analytical Procedures for Quality of mRNA Vaccines and Therapeutics (Draft Guidelines: 3rd Edition). 2024 Aug 2. at https://www.uspnf.com/notices/analytical-procedures-mrna-vaccines-20240802 .)
113. Wiseman D, McKernan K, Gutschi L, Rose J. Comments on USP Draft Guidelines: Analytical Procedures for mRNA Vaccine Quality, 2nd edition v2. ResearchGate 2023 July 26. Epub Jul 26 http://doi.org/10.13140/RG.2.2.33586.99526
114. EMA. Draft guideline on the quality aspects of mRNA vaccines. Committee for Medicinal Products for Human Use (CHMP) EMA/CHMP/BWP/82416/2025. 2025 March 27. at https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-quality-aspects-mrna-vaccines_en.pdf .)
115. WHO. WHO Study Group on Cell Substrates for Production of Biologicals 2007 June 11-12. at https://cdn.who.int/media/docs/default-source/biologicals/cell-substrates/cells.final.mtgrep.ik.26_sep_07.pdf?sfvrsn=3db7d37a_3&download=true .)
116. EMA. AskEMA - Response to David Wiseman - ASK-154880 - SV40 sequence in Covid19 plasmid. 2023 Nov 27.
117. Wang Z, Troilo PJ, Wang X, et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther 2004; 11:711-21. Epub http://doi.org/10.1038/sj.gt.3302213
118. Canada H. ATIP Release Package HC-A-2023-001013 - 2024-08-09 to Noe Chartiers. 2024 August. at https://www.researchgate.net/publication/386986657_ReleasePackage_HC-A-2023-001013_-_2024-08-09_OCR_Noe_Chartiers_August_2024 .)
119. Drayman N, Ben-Nun-Shaul O, Butin-Israeli V, et al. p53 elevation in human cells halt SV40 infection by inhibiting T-ag expression. Oncotarget 2016; 7:52643-60. Epub http://doi.org/10.18632/oncotarget.10769
120. Klinman DM, Klaschik S, Tross D, Shirota H, Steinhagen F. FDA guidance on prophylactic DNA vaccines: analysis and recommendations. Vaccine 2010; 28:2801-5. Epub 20091124 http://doi.org/10.1016/j.vaccine.2009.11.025
121. Valera A, Perales JC, Hatzoglou M, Bosch F. Expression of the neomycin-resistance (neo) gene induces alterations in gene expression and metabolism. Hum Gene Ther 1994; 5:449-56. Epub http://doi.org/10.1089/hum.1994.5.4-449
122. Poulin DL, DeCaprio JA. Is there a role for SV40 in human cancer? J Clin Oncol 2006; 24:4356-65. Epub http://doi.org/10.1200/JCO.2005.03.7101
123. Butel JS. SV40 large T-antigen: dual oncogene. Cancer Surv 1986; 5:343-65. Epub ,
124. Harford JB. A Second Career for p53 as A Broad-Spectrum Antiviral? Viruses 2023; 15. Epub 20231203 http://doi.org/10.3390/v15122377
125. EMA. EMA/895061/2022 Assessment report Invented name: COMIRNATY Original/Omicron BA.4-5 Committee for Medicinal Products for Human Use (CHMP). 2022 Sep 12. at https://www.ema.europa.eu/en/documents/variation-report/comirnaty-h-c-005735-ii-0143-epar-assessment-report-variation_en.pdf .)
126. Health Canada. Health Canada ATIP: Response to Post-Decision Letter Dated: October 20, 2023. Release Package - HC A-2024-000097 - 2024-08-22-2. 2024. at https://www.researchgate.net/publication/395401666_Release_Package_-_HC_A-2024-000097_-_2024-08-22-2 .)
127. Prasad TK, Rao NM. The role of plasmid constructs containing the SV40 DNA nuclear-targeting sequence in cationic lipid-mediated DNA delivery. Cell Mol Biol Lett 2005; 10:203-15. Epub ,
128. Packer M, Gyawali D, Yerabolu R, Schariter J, White P. A novel mechanism for the loss of mRNA activity in lipid nanoparticle delivery systems. Nat Commun 2021; 12:6777. Epub 20211122 http://doi.org/10.1038/s41467-021-26926-0
129. Moderna. Moderna Science & Technology Day: Presentations. 2022 May 17. at https://s29.q4cdn.com/435878511/files/doc_presentations/2022/05/Science-Day-2022-Master-Slides-FINAL-(05.17_7am).pdf .)
130. Moderna. Annual Science Day. 2020 June 2. at https://s29.q4cdn.com/435878511/files/doc_presentations/2020/06/02/Science-Day-Master-Final-(06.02.20)_updated-from-Ed-(1).pdf .)
131. Moderna. NON-GLP FINAL REPORT AMENDMENT NO. 01 Test Facility Study No. 5002121 A Single Dose Intramuscular Injection Tissue Distribution Study of mRNA-1647 in Male Sprague-Dawley Rats (Charles River) obtained via FOIA. 2017. at https://www.judicialwatch.org/documents/jw-v-hhs-biodistribution-prod-4-02418-pgs-370-649/ .)
132. Jörgensen AM, Wibel R, Bernkop-Schnürch A. Biodegradable Cationic and Ionizable Cationic Lipids: A Roadmap for Safer Pharmaceutical Excipients. Small 2023; 19:2206968. Epub http://doi.org/https://doi.org/10.1002/smll.202206968
133. Vijayraghavan S, Saini N. Aldehyde-Associated Mutagenesis─Current State of Knowledge. Chem Res Toxicol 2023; 36:983-1001. Epub http://doi.org/10.1021/acs.chemrestox.3c00045
134. Housh K, Jha JS, Haldar T, et al. Formation and repair of unavoidable, endogenous interstrand cross-links in cellular DNA. DNA Repair (Amst) 2021; 98:103029. Epub 20201224 http://doi.org/10.1016/j.dnarep.2020.103029
135. FDA. Guidance for Industry S2B Genotoxicity: A Standard Battery for Genotoxicity Testing of Pharmaceuticals. 1997 Nov 21. at https://www.fda.gov/media/71971/download .)
136. EMA. Assessment report: COVID-19 Vaccine Moderna 2021 11 Mar. at https://www.ema.europa.eu/en/documents/assessment-report/spikevax-previously-covid-19-vaccine-moderna-epar-public-assessment-report_en.pdf .)
137. Panzner S, Sahin U, Krijger J-J, et al., inventors; BioNtech SE, assignee. LNP compositions comprising RNA and methods for preparing, storing and using the same. Patent application US20230414747A1 Filed Nov 15 20212023 Dec 28.
138. Wiseman D, Rose, J, Guetzkow, H, Seligmann H. Why limit contraindication to Janssen? Using same criteria revisit EUA/BLA for all C19 quasi-vaccines. Transparency: Emergency ACIP Meeting Dec 16 2021. CDC-2021-0133. ResearchGate 2021 Dec 23. Epub http://doi.org/https://dx.doi.org/10.13140/RG.2.2.32783.51368
139. Hassett KJ, Benenato KE, Jacquinet E, et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol Ther Nucleic Acids 2019; 15:1-11. Epub 20190207 http://doi.org/10.1016/j.omtn.2019.01.013
140. Henderson MI, Eygeris Y, Jozic A, Herrera M, Sahay G. Leveraging Biological Buffers for Efficient Messenger RNA Delivery via Lipid Nanoparticles. Mol Pharm 2022; 19:4275-85. Epub 20220921 http://doi.org/10.1021/acs.molpharmaceut.2c00587
141. FDA. FDA Authorizes Pfizer-BioNTech COVID-19 Vaccine for Emergency Use in Children 5 through 11 Years of Age. 2021 Oct 29. Epub , https://www.fda.gov/news-events/press-announcements/fda-authorizes-pfizer-biontech-covid-19-vaccine-emergency-use-children-5-through-11-years-age
142. Wiseman D. ACIP October 19-20-2022. BA4/5 bivalent quasi-vaccines in yet younger children: Further erosion of scientific and ethical standards. Written and Oral Comments. Research Gate 2022 Oct. Epub Oct 19 http://doi.org/www.regulations.gov/comment/CDC-2022-0111-126227
143. Wiseman D, Guetzkow, J, Pantazatos, S, Rose, J, Seligmann, H. National Academies Committee on Review of Relevant Literature Regarding Adverse Events Associated with Vaccines March 30 2023: Written material accompanying oral remarks. Research Gate 2023. Epub April 3 http://doi.org/10.13140/RG.2.2.27009.74089
144. FDA. EUA Revision October 12 - Pfizer Bivalent Booster Doses - 5-11 years. 2022 October 12. at https://www.fda.gov/media/150386/download .)
145. FDA. EUA Revision August 31 - Moderna Bivalent Booster Doses. 2022 August 31. at https://www.fda.gov/media/144636/download .)
146. Tegally H, Moir M, Everatt J, et al. Continued Emergence and Evolution of Omicron in South Africa: New BA.4 and BA.5 lineages. medRxiv 2022:2022.05.01.22274406. Epub May 2 http://doi.org/10.1101/2022.05.01.22274406
147. Wiseman D. BA4/5 bivalent quasi-vaccines: Further relaxation of FDA standards, manufacturing changes and novel spike protein heterotrimers. Written comments to CDC ACIP meeting of September 1 2022. CDC-2022-0103-0049. Research Gate 2022 Sep 1. Epub http://doi.org/10.13140/RG.2.2.25633.28007
148. Wagenhäuser I, Reusch J, Gabel A, et al. Bivalent BNT162b2mRNA original/Omicron BA.4-5 booster vaccination: adverse reactions and inability to work compared to the monovalent COVID-19 booster. medRxiv 2022:2022.11.07.22281982. Epub Nov 8 http://doi.org/10.1101/2022.11.07.22281982
149. FDA. Emergency Use Authorization for Vaccines to Prevent COVID-19 Guidance for Industry. This document supersedes the guidance of the same title issued on
May 25, 2021. 2022 Mar 31. at https://www.fda.gov/media/142749/download .)
150. Wiseman D. Covid-19 vaccine safety, bivalent primary series, future directions. Written comments submitted to CDC- ACIP February 24 2023. CDC-2023-0007-0496. Research Gate 2023 Feb 24. Epub http://doi.org/10.13140/RG.2.2.25839.10404
151. FDA. Summary Basis for Regulatory Action - Moderna Spikevax. 2022 Jan 30. at https://www.fda.gov/media/155931/download .)
152. Moderna. 2.4 Nonclinical Overview, Obtained via FOIA by Judicial Watch, Inc. (Three versions)
31pp at FDA-CBER-2021-4379-0001130
27pp at FDA-CBER-2021-4379-0001494
32pp at FDA-CBER-2021-4379-0001462. 2021. at at 671, 311. 338 www.judicialwatch.org/documents/jw-v-hhs-biodistribution-prod-4-02418/ .)
153. Hassett KJ, Higgins J, Woods A, et al. Impact of lipid nanoparticle size on mRNA vaccine immunogenicity. Journal of Controlled Release 2021; 335:237-46. Epub http://doi.org/https://doi.org/10.1016/j.jconrel.2021.05.021
154. Moderna. 2.6.4 Pharmacokinetics Written Summary. Obtained by FOIA. 2020. at https://www.judicialwatch.org/documents/jw-v-hhs-biodistribution-prod-4-02418-pgs-291-303/ .)
155. Takayama K. In Vitro and Animal Models for SARS-CoV-2 research. Trends Pharmacol Sci 2020. Epub 2020/06/20 http://doi.org/10.1016/j.tips.2020.05.005
156. Jiang H, Hyddmark EMV, Gordon S, et al. Development of humanized ACE2 mouse and rat models for COVID-19 research. The FASEB Journal 2022; 36. Epub http://doi.org/https://doi.org/10.1096/fasebj.2022.36.S1.R2068
157. Brooke GN, Prischi F. Structural and functional modelling of SARS-CoV-2 entry in animal models. Sci Rep 2020; 10:15917. Epub 20200928 http://doi.org/10.1038/s41598-020-72528-z
158. Bao L, Deng W, Huang B, et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 2020; 583:830-3. Epub http://doi.org/10.1038/s41586-020-2312-y
159. Winkler ES, Bailey AL, Kafai NM, et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat Immunol 2020; 21:1327-35. Epub http://doi.org/10.1038/s41590-020-0778-2
160. Pfizer. BNT162b2 Module 2.4. Nonclinical Overview. Document released pursuant to FOIA. 2021 Feb 8. at https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M2_24_nonclinical-overview.pdf .)
161. Kirschman JL, Bhosle S, Vanover D, et al. Characterizing exogenous mRNA delivery, trafficking, cytoplasmic release and RNA-protein correlations at the level of single cells. Nucleic Acids Res 2017; 45:e113. Epub http://doi.org/10.1093/nar/gkx290
162. Zhang H, Barz M. Investigating the stability of RNA-lipid nanoparticles in biological fluids: Unveiling its crucial role for understanding LNP performance. Journal of Controlled Release 2025; 381:113559. Epub http://doi.org/https://doi.org/10.1016/j.jconrel.2025.02.055
163. Gao F, Mallajoysula V, Arunachalam PS, et al. Spheromers reveal robust T cell responses to the Pfizer/BioNTech vaccine and attenuated peripheral CD8(+) T cell responses post SARS-CoV-2 infection. Immunity 2023. Epub 20230316 http://doi.org/10.1016/j.immuni.2023.03.005
164. Pfizer. BNT162b2 Module 2.6.4 Pharmacokinetics Written Summary. Released under FOIA. 2021 February 8. at https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M2_26_pharmkin-written-summary.pdf .)
165. Pfizer. BNT162b2 MODULE 2.6.5. PHARMACOKINETICS TABULATED SUMMARY. Released under FOIA. 2021 January 21. at https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M2_26_pharmkin-tabulated-summary.pdf .)
166. Pfizer. PFIZER-BIONTECH COVID-19 VACCINE (BNT162, PF-07302048) VACCINES AND RELATED BIOLOGICAL PRODUCTS ADVISORY COMMITTEE
BRIEFING DOCUMENT MEETING DATE: 10 December 2020. 2020. (Accessed Oct 25, 2021, at https://www.fda.gov/media/144246/download .)
167. FDA. Briefing Document Pfizer-BioNTech COVID-19 Vaccine. 2020 Dec 10. at https://www.fda.gov/media/144245/download .)
168. FDA. Emergency Use Authorization (EUA) for an Unapproved Product Review Memorandum (Pfizer-BioNTech COVID-19 Vaccine/ BNT162b2). 2020 Dec 11. at https://www.fda.gov/media/144416/download .)
169. Moderna. MRNA-1273 SPONSOR BRIEFING DOCUMENT VRBPAC. 2020 Dec 17. at https://www.fda.gov/media/144452/download
https://www.fda.gov/media/144453/download .)
170. FDA. Vaccines and Related Biological Products Advisory Committee Meeting FDA Briefing Document Moderna COVID-19 Vaccine. 2020 Dec 17. at https://www.fda.gov/media/144434/download .)
171. FDA. Emergency Use Authorization (EUA) for an Unapproved Product Review Memorandum: Moderna. 2020 Nov 30. at https://www.fda.gov/media/144673/download .)
172. WHO. Guidelines on the non-clinical evaluation of vaccine adjuvants and adjuvanted vaccines, Annex 2, TRS No 987. 2014. at https://www.who.int/publications/m/item/nonclinical-evaluation-of-vaccine-adjuvants-and-adjuvanted-vaccines-annex-2-trs-no-987 .)
173. Vervaeke P, Borgos SE, Sanders NN, Combes F. Regulatory guidelines and preclinical tools to study the biodistribution of RNA therapeutics. Adv Drug Deliv Rev 2022; 184:114236. Epub 2022/03/31 http://doi.org/10.1016/j.addr.2022.114236
174. Moderna. 2.6.1 Introduction, Nonclinical Development Program for mRNA-1273, obtained via FOIA. 2020. at https://www.judicialwatch.org/documents/jw-v-hhs-biodistribution-prod-4-02418/ .)
175. Orlandini von Niessen AG, Poleganov MA, Rechner C, et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3' UTRs Identified by Cellular Library Screening. Mol Ther 2019; 27:824-36. Epub 2019/01/15 http://doi.org/10.1016/j.ymthe.2018.12.011
176. Blumberg A, Zhao Y, Huang YF, et al. Characterizing RNA stability genome-wide through combined analysis of PRO-seq and RNA-seq data. BMC Biol 2021; 19:30. Epub 20210215 http://doi.org/10.1186/s12915-021-00949-x
177. WHO. Annex 1: WHO guidelines on nonclinical evaluation of vaccines WHO Technical Report Series, No. 927. 2005. at https://www.who.int/publications/m/item/nonclinical-evaluation-of-vaccines-annex-1-trs-no-927 .)
178. Moderna. 2.6.5 Pharmacokinetics Tabulated Summary, obtained by FOIA. 2020. at https://www.judicialwatch.org/documents/jw-v-hhs-biodistribution-prod-4-02418/ .)
179. EMEA. GUIDELINE ON EXCIPIENTS IN THE DOSSIER FOR APPLICATION FOR MARKETING AUTHORISATION OF A MEDICINAL PRODUCT 2007 June 19. at https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-excipients-dossier-application-marketing-authorisation-medicinal-product-revision-2_en.pdf .)
180. Hemmrich E, McNeil S. Active ingredient vs excipient debate for nanomedicines. Nat Nanotechnol 2023. Epub 20230427 http://doi.org/10.1038/s41565-023-01371-w
181. Peden K. Considerations for the Quality, Safety and Efficacy of Prophylactic Lipid Nanoparticle mRNA Vaccines. Public Workshop on FDA Guidance to Industry on Nanomaterials. 2022 Oct 11. at https://www.fda.gov/media/166986/download
https://youtu.be/xszK_ug7QEw?t=1684 .)
182. EMEA. GUIDELINE ON ADJUVANTS IN VACCINES FOR HUMAN USE. 2005. at https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-adjuvants-vaccines-human-use-see-also-explanatory-note_en.pdf .)
183. FDA. Guideline for Industry Specific Aspects of Regulatory Genotoxicity Tests for Pharmaceuticals. 1996 Apr 24. at https://www.fda.gov/media/71959/download .)
184. FDA. M7(R2) Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk Guidance for Industry. 2023 July 23. at https://www.fda.gov/media/170461/download .)
185. FDA. M7(R2) Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk Questions and Answers Guidance for Industry. 2023 Jul 23. at https://www.fda.gov/media/170460/download .)
186. Vanderkerken K, Vanparys P, Verschaeve L, Kirsch-Volders M. The mouse bone marrow micronucleus assay can be used to distinguish aneugens from clastogens. Mutagenesis 1989; 4:6-11. Epub http://doi.org/10.1093/mutage/4.1.6
187. FDA. Guidance for Industry. S2(R1) Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use. 2012 June 7. at https://www.fda.gov/media/71980/download .)
188. FDA. Guidance for Industry and Review Staff Recommended Approaches to Integration of Genetic Toxicology Study Results. 2006 Jan 4. at https://www.fda.gov/media/72266/download .)
189. Grawé J, Zetterberg G, Amnéus H. DNA content determination of micronucleated polychromatic erythrocytes induced by clastogens and spindle poisons in mouse bone marrow and peripheral blood. Mutagenesis 1993; 8:249-55. Epub http://doi.org/10.1093/mutage/8.3.249
190. FDA. Redbook 2000: Mammalian Erythrocyte Micronucleus Test Toxicological Principles for the Safety Assessment of Food Ingredients Redbook Chapter IV.C.1.d. Mammalian Erythrocyte Micronucleus Test. 2000 July. at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/redbook-2000-ivc1d-mammalian-erythrocyte-micronucleus-test .)
191. Mortelmans K, Zeiger E. The Ames Salmonella/microsome mutagenicity assay. Mutat Res 2000; 455:29-60. Epub http://doi.org/10.1016/s0027-5107(00)00064-6
192. OECD. Organisation for Economic Co-operation and Development. ENVIRONMENT DIRECTORATE JOINT MEETING OF THE CHEMICALS COMMITTEE AND THE WORKING PARTY ON CHEMICALS, PESTICIDES AND BIOTECHNOLOGY. The in vivo erythrocyte Pig-a gene mutation assay – Part 1 – Detailed Review Paper and Retrospective Performance Assessment. 2020 16 Jul. (Accessed Nov 30, 2021, at https://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/jm/mono(2020)6&doclanguage=en .)
193. FDA. S1A The Need for Long-term Rodent Carcinogenicity Studies of Pharmaceuticals. 1996 March 1. at https://www.fda.gov/media/71921/download .)
194. FDA. Guidance for Industry S1B Testing for Carcinogenicity of Pharmaceuticals. 1997 July. at https://www.fda.gov/media/71935/download .)
195. FDA. Spikevax Package Insert 2024-2025 Formula. 2024 August 22. at https://www.fda.gov/media/155675/download?attachment .)
196. Angues RV, Bustos YP, Angues RV, Bustos YP. SARS-CoV-2 Vaccination and the Multi-Hit Hypothesis of Oncogenesis. Cureus 2023; 15. Epub Dec 17 http://doi.org/10.7759/cureus.50703
197. FDA. Drug Products, Including Biological Products, that Contain Nanomaterials Guidance for Industry. 2022 April. at https://www.fda.gov/media/157812/download .)
198. FDA. Guidance for Industry Considering Whether an FDA-Regulated Product Involves the Application of Nanotechnology. 2014 June. at https://www.fda.gov/media/88423/download .)
199. Pfizer. Pfizer Documents released under FOIA to Judicial Watch Feb 28 2022. at https://www.judicialwatch.org/wp-content/uploads/2022/03/JW-v-HHS-prod-3-02418.pdf
https://www.judicialwatch.org/documents/jw-v-hhs-fda-pfizer-biontech-vaccine-prod-3-02418/ .)
200. EMA. S 2 B: Note for guidance on genotoxicity: A standard battery for genotoxicity testing of pharmaceuticals CPMP/ICH/174/95. 1998 Mar 1. at https://www.ema.europa.eu/en/documents/scientific-guideline/s-2-b-note-guidance-genotoxicity-standard-battery-genotoxicity-testing-pharmaceuticals_en.pdf .)
201. Marks P. Testimony before the Select Subcommittee on the Coronavirus Pandemic Committee on Oversight and Accountability U.S. House of Representatives. 2024 Feb 15. at https://oversight.house.gov/hearing/assessing-americas-vaccine-safety-systemspart-1/
https://www.youtube.com/watch?v=c5hYh5XO7qY&t .)
202. Stieber Z. Epoch Times. CDC Finds Hundreds of Safety Signals for Pfizer, Moderna COVID Vaccines. 2023 Jan 3. at https://www.theepochtimes.com/health/exclusive-cdc-finds-hundreds-of-safety-signals-for-pfizer-and-moderna-covid-19-vaccines-4956733 .)
203. FDA. FOIA disclosure to ICAN regarding EBDM and PRR VAERS analyses. 2025 Jan 22. at https://filedev0128.s3.us-east-1.amazonaws.com/ratio-tables/cdc-proportional-reporting-ratio-tables.zip
https://www.fda.gov/media/184988/download .)
204. Wiseman D. Signal loss by truancy, masking, and filtering, and underestimation of potential risks and suspected adverse reactions in the Disproportionality Signal Analyses of VAERS data associated with COVID-19 pro-vaccines. Research Gate 2025. Epub Sept 9 http://doi.org/http://dx.doi.org/10.13140/RG.2.2.16568.40961
205. FEDERAL FOOD, DRUG, AND COSMETIC ACT [As Amended Through P.L. 117–328, Enacted December 29, 2022]. https://www.govinfo.gov/content/pkg/COMPS-973/pdf/COMPS-973.pdf
206. Alegria C, Nunes Y. UK - Death and Disability Trends for Malignant Neoplasms, Ages 15-44. ResearchGate 2024. Epub http://doi.org/10.13140/RG.2.2.34374.45123
207. Alegria C, Wiseman D, Nunes Y. US -Death Trends for Neoplasms ICD codes: C00-D48, Ages 15-44. ResearchGate 2024. Epub Mar 11 http://doi.org/10.13140/RG.2.2.16068.64645
208. Wiseman D. COVID-19 era cancers: trends and challenges. Testimony to THE SENATE OF TEXAS COMMITTEE ON HEALTH AND HUMAN SERVICES. 2024 May 14. at https://tlcsenate.granicus.com/MediaPlayer.php?clip_id=18499 .)
209. Mulroney TE, Pöyry T, Yam-Puc J, et al. (N)1-methylpseudouridylation of mRNA causes +1 ribosomal frameshifting. Nature 2023. Epub Dec 6 http://doi.org/10.1038/s41586-023-06800-3
210. University of Cambridge. Researchers redesign future mRNA therapeutics to prevent potentially harmful immune responses. 2023 Dec 6. at https://www.cam.ac.uk/research/news/researchers-redesign-future-mrna-therapeutics-to-prevent-potentially-harmful-immune-responses .)
211. Peabody DS. Translation initiation at non-AUG triplets in mammalian cells. J Biol Chem 1989; 264:5031-5. Epub ,
212. Autelitano DJ, Rajic A, Smith AI, et al. The cryptome: a subset of the proteome, comprising cryptic peptides with distinct bioactivities. Drug Discov Today 2006; 11:306-14. Epub http://doi.org/10.1016/j.drudis.2006.02.003
213. Cordes J, Zhao S, Engel CM, Stingele J. Cellular responses to RNA damage. Cell 2025; 188:885-900. Epub http://doi.org/10.1016/j.cell.2025.01.005
214. WHO. WHO Expert Committee on Biological Standardization. Seventy-fourth report: 74th report: WHO TRS N°1039. 2022 Apr 12. at https://www.who.int/publications/i/item/9789240046870 .)
215. Wiseman D, Seligmann, H, Pantazatos, SP. Covid-19 gene therapy vaccines: Why no review by FDA’s Office of Tissues and Advanced Therapies (OTAT) and Cell Therapy Gene Therapy Advisory Committee (CTGTAC) Written comments submitted to FDA- CTGTAC Meeting June 10th 2022 FDA-2022-N-0470. Research Gate 2022 June 2. Epub Nov 21 http://doi.org/10.13140/RG.2.2.29921.68964
216. FDA. Transfer of Therapeutic Biological Products to the Center for Drug Evaluation and Research. 2022 March 7. (Accessed September 12, at https://www.fda.gov/combination-products/jurisdictional-information/transfer-therapeutic-biological-products-center-drug-evaluation-and-research .)
217. FDA. Food and Drug Administration. Long Term Follow-up After Administration of Human Gene Therapy Products. Guidance for Industry. FDA-2018-D-2173. 2020. (Accessed July 13, 2021, at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/long-term-follow-after-administration-human-gene-therapy-products
https://www.fda.gov/media/113768/download .)
218. FDA. Guidance for Industry Preclinical Assessment of Investigational Cellular and Gene Therapy Products 2013. (Accessed March 30, 2022, at https://www.fda.gov/media/87564/download .)
219. Moderna. QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the quarterly period ended June 30, 2020. 2020 Aug 6. (Accessed July 22, 2021, at https://www.sec.gov/Archives/edgar/data/1682852/000168285220000017/mrna-20200630.htm .)
220. CDC. Safety of COVID-19 Vaccines. 2023 Mar 7. (Accessed July 7, 2023, at https://www.cdc.gov/coronavirus/2019-ncov/vaccines/safety/safety-of-vaccines.html .)
221. Eisenstein M. Vaccination rates are falling, and its not just the COVID-19 vaccine that people are refusing. Nature 2022; 612:S44-s6. Epub Dec 19 http://doi.org/10.1038/d41586-022-04341-9
222. Gutschi LM SF. Rethinking Lipid Nanoparticles (LNPs): Biological Activity, Safety, and Policy Implications for modRNA Therapeutics Key Message. ResearchGate 2025. Epub September 12 http://doi.org/http://dx.doi.org/10.13140/RG.2.2.34138.61127
223. Kingwell K. COVID vaccines: “We flew the aeroplane while we were still building it”. Nature Reviews Drug Disc 2022. Epub Nov 11 http://doi.org/doi.org/10.1038/d41573-022-00191-2
